
GeneMap '98 Features 30,000 Human Genes
Cn3D 2.0 Enhances Structural Analysis
PHI-BLAST: Motif-Constrained Sequence
Similarity Searches
Submitting Identical Sequences
from Multiple Sources to GenBank
Authorin No Longer Accepted by GenBank
Using the Genes and Disease
Web Site
Organism Filters and Greater
Flexibility Added to BLAST
dbSNP: A Database of Single Nucleotide
Polymorphisms
GenBank Tops Two Billion Base Pairs as
NCBI Marks 10th Anniversary
Selected Recent Publications by NCBI Staff
Masthead
A complete human gene map will be essential to progress toward a deeper understanding of human biology and disease. Complete genomic sequences now exist for many organisms with genome sizes on the order of 20 megabases, but the sequencing of the 3-gigabase human genome will require a number of years to complete. Meanwhile, a human transcript map can serve as a valuable sequencing and mapping framework. An international consortium was formed in 1994 to construct such a map by determining the locations of expressed sequence tags (ESTs) relative to a framework of well-characterized genetic markers. The first map to result from this effort was the Gene Map of the Human Genome, which appeared in 1996 and included 16,354 gene-based markers.1 The successor to this 1996 transcript map, GeneMap ’98,2 features 30,261 unique gene loci, or about half of the 60,000 to 80,000 genes thought to be contained in the human genome.
Building GeneMap ’98: Radiation Hybrid Mapping
The process of constructing GeneMap ’98 involved the mapping of short (typically, only a few hundred base pairs), unique DNA sequences, termed sequence tagged sites (STSs), within a framework of well-characterized genetic markers developed by the Genethon Corporation. The technique used to locate the STSs within the framework of genetic markers is known as radiation hybrid (RH) mapping.
Radiation hybrid mapping employs a panel of human-on-hamster hybrid cell lines, each containing its native hamster genome as well as a random assortment of human chromosomal fragments. These chromosomal fragments are produced by X-ray irradiation of human genomic DNA to produce random, radiation-induced breaks in the chromosomes. Two such hybrid cell panels have been employed to create GeneMap ’98: the GeneBridge 4 (GB4) panel and the Stanford G3. The two RH panels complement one another in that GB4 provides greater long-range continuity, whereas G3 provides increased map resolution.
The location of an STS is determined by performing a set of polymerase chain reaction (PCR) assays using the primer pair associated with the STS in question and the DNA isolated from each of the various cell lines constituting a radiation hybrid panel. If the target STS is present within the human DNA fragments isolated from a particular cell line, a PCR product of the correct length is observed. In this case, the marker is said to be "retained" within the DNA of the cell line yielding the correct PCR product.
The STS is mapped by performing a statistical comparison of its pattern of retention within the cell lines constituting the RH panel with the retention patterns arising from other STSs.
Reading the Map
A row of hyperlinks running across the top of the GeneMap ’98 page provides a path to each of the 22 human autosomal chromosomes as well as to the X chromosome. The Y chromosome is not represented because the GB4 RH panel was constructed using a female (XX) genome. Following the link to chromosome 22, for instance, leads to an ideogram of chromosome 22 with labeled G-bands as shown in Figure 1. This chromosomal view will be referred to as the "map view." Aligned to the left of the chromosome graphic are depictions of the G3 RH, the GB4 RH, and the Genethon Genetic maps, as well as a histogram of gene density along the chromosome. The common framework markers that have been used to integrate these three independent maps are connected with lines in the map view and divide the chromosomes into discrete intervals.
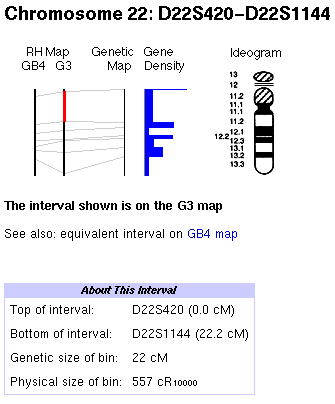
Figure 1. Graphical overview of chromosome 22 in GeneMap ’98.
The initial interval displayed when first selecting a chromosome
is the p-terminal (upper) interval on the GB4 RH map. To display
the second interval on the G3 map, shown in Figure 1, click on
the G3 line between the first and second framework lines. The
selected interval is highlighted in the graphical display by a
thick line (red in the Web graphic), as shown in the figure.
Beneath the chromosomal ideogram is a table providing detailed information on the mapped sites within the interval. The table consists of a list of markers given in the order in which they are found within the selected interval. The location of each marker that appears in the Genethon genetic map is given in centimorgans (cM). An associated LOD (log odds) score provides a measure of the confidence with which the location of the marker is known. Hyperlinks to more information on particular markers and short descriptions of the markers are also included.
Application
GeneMap ’98 will provide a scaffold on which to mount the large-scale sequencing data being generated daily as the sequencing of the human genome progresses. The map will also accelerate the pace of the discovery of human disease genes by positional cloning. In the year following the publication the 1996 Gene Map of the Human Genome, for instance, the isolation of 16 genes by positional cloning techniques was reported. Of these 16 genes, 7 had already been isolated as ESTs and mapped at the time of their cloning. GeneMap ’98 now includes mapping information for 11 of these 16 genes.
GeneMap ’98 can be reached by following the Gene Map of the Human Genome link from NCBI’s home page. A link to the 1996 human transcript map is provided on the GeneMap ’98 page. The GeneMap ’98 map data files are available at ftp://ncbi.nlm.nih.gov/repository/genemap/Oct1998/.
Notes
1 Schuler, GD, et al. A gene map of the human genome. Science 274:540–6, 1996.
2 Deloukas, P, et al. A physical map of 30,000 human genes. Science 744–6, 1998.
Cn3D is a molecular structure viewer developed at NCBI to allow users to view protein and nucleic acid structures from the Molecular Modeling Database (MMDB).1 Configured as a Web browser helper-application, Cn3D can be invoked automatically when an MMDB format molecular structure is downloaded. Cn3D 2.0 provides major enhancements over version 1.0 by integrating the display of the 3D structure of a protein or nucleic acid with a display of its primary sequence. The program accomplishes this by making use of separate sequence and structure windows that are linked in such a way that regions of primary sequence can be visually correlated with regions of structure.
Cn3D 2.0 also uses the linkage between the structural and the sequence windows to provide powerful structural analysis capabilities. A BLAST-based alignment feature of the sequence window aligns imported protein sequences to a structurally anchored master sequence. In this manner, a protein sequence for which structural information is lacking can be mapped to a known model structure if sequence homology between the query sequence and the model exists. Precomputed alignments may also be imported.
Another improvement is Cn3D 2.0’s ability to load and display the 3D alignments of protein domains created and maintained at NCBI using the VAST (Vector Alignment Search Tool) program. In this case, the sequence window displays the sequence alignment that is implied by the VAST structural alignment. These features of Cn3D are highlighted in the examples below.
Mapping a Sequence to a Model Structure
Figure 1 shows a Cn3D rendering of the structure of the soluble domain of oxidized cytochrome B5 from Bos taurus.2 Within the structure window, the protein is rendered as a "tubular" model, whereas the prosthetic heme group is rendered as a "space-filling" model. The sequence window contains two cytochrome B5 sequences. The first sequence is the master sequence and corresponds to the protein structure visible in the structure window. The second sequence is that of cytochrome B5 of the distant eukaryote, Saccharomyces cerevisiae.3 Although the structure of the yeast protein is unknown, its sequence has been imported into the sequence window of Cn3D and automatically aligned with the Bos taurus sequence. Because the alignment is good, it is possible for Cn3D to "map" the yeast sequence onto the Bos taurus structure. This has been visualized by coloring the structure by Protein to produce a solid dark tubular structure and then choosing Show Substitutions from the Alignment menu in the sequence window. The resulting rendering in the structure window shows a dark tubular trace where the two sequences are identical and a lighter, highlighted trace where they differ. This highlighting is reflected in the sequence window by the light shading of nonconserved residues in the 1CYO sequence. In this manner, the sequence differences between the two cytochrome B5 proteins have been mapped onto the structure of the Bos taurus protein.
The mapping reveals that a conspicuous run of conserved residues in the sequence alignment, "EHPGG" in the two sequences, forms a loop in the structure of the Bos taurus cytochrome. This loop contains the leftmost of two conserved histidines, which are seen "kissing" the central iron of the heme prosthetic group from opposite sides.
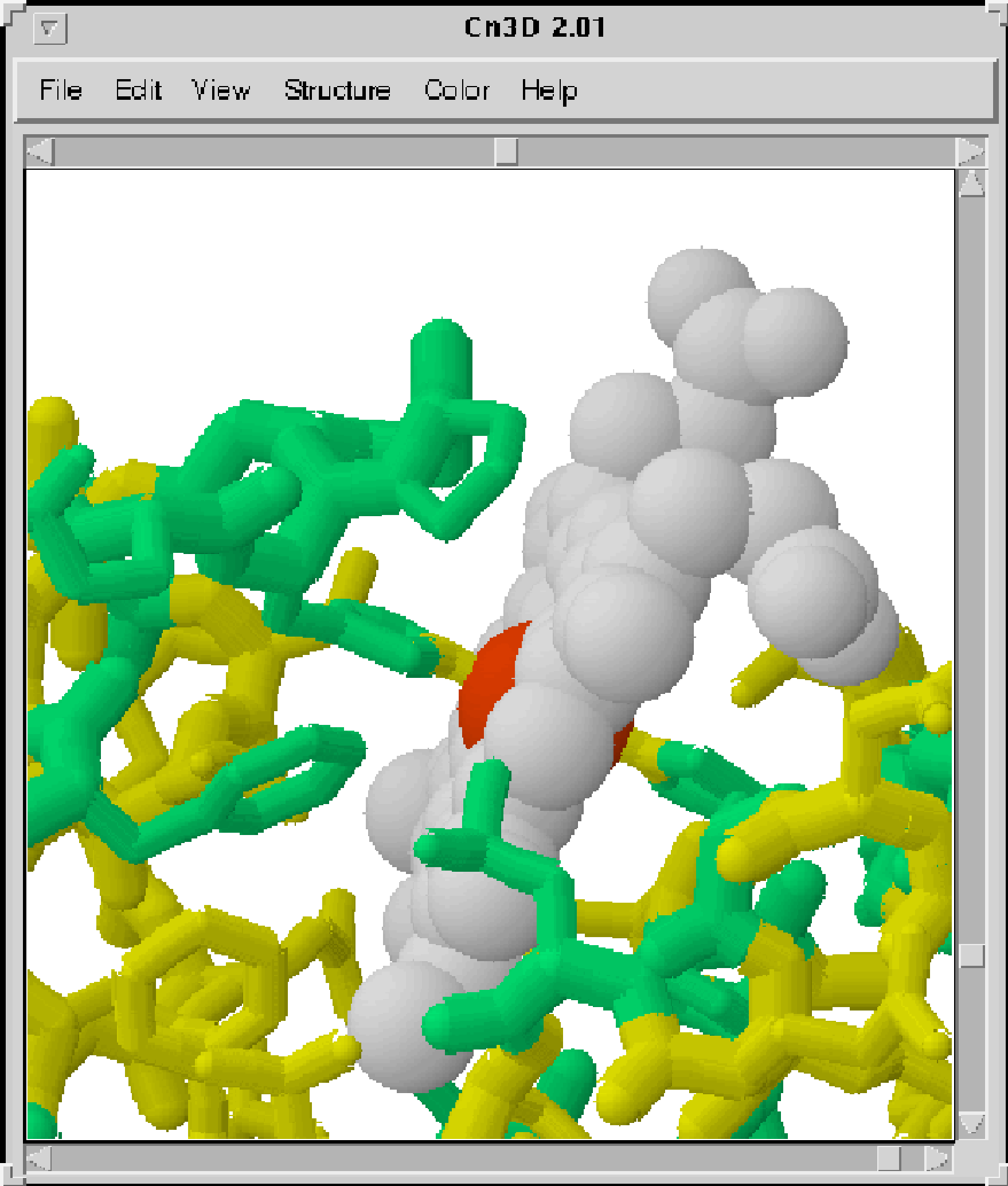

Figure 1. Close-up of the
heme-binding pocket of bovine cytochrome B (PDB code 1CYO)
showing
the interaction of conserved residues with the heme (space-filled
structure) group. The alignment of
the 1CYO sequence with a homologous sequence from S.
cerevisiae is shown below.
Viewing a VAST Protein Alignment
A new feature of Cn3D 2.0 is its ability to display the protein domain structural alignments constituting the NCBI VAST database. Figure 2 shows a Cn3D structure window, which displays a rendering of the VAST alignment of two 5' to 3' exonucleases, 1TFR4 and 1EXN5 from bacteriophages T4 and T5, respectively. The proteins are depicted as two partially superimposed alpha-carbon traces. The trace for 1TFR is colored green (lighter shade), and that for 1EXN, blue (darker shade). Two catalytically critical magnesium ions, seen in the 1TFR structure, are visible as dark spheres.

Figure 2. Superimposed
alpha-carbon traces for 1TFR (light trace) and 1EXN
(darker trace) resulting from a VAST-generated structural
alignment.
Figure 2 highlights an instance in which one crystal structure complements a second by providing a model for regions that are disordered in the second structure. The region of interest is the central region in the structure window. The residues in this region form a helical arch in the 1EXN structure (darker trace), which passes over the central cleft of the enzyme core. Note the two abnormal termini on either side of the cleft in the light-colored 1TFR trace where the disordered segment lies. It has been suggested that single-stranded DNA passes through this arch as part of the enzyme’s catalytic mechanism.5 From the alignment it may be hypothesized that the disordered segment of amino acids in 1TFR also forms an arch similar to that in 1EXN.
Notes
1 Hogue, CW, H Ohkawa, and SH Bryant. A dynamic look at structures: WWW-Entrez and the Molecular Modeling Database. Trends Biochem Sci 21(6):226–9, 1996.
2 Mathews, FS, P Argos, and M Levine. The structure of cytochrome b 5 at 2.0 Angstrom resolution. Cold Spring Harb Symp Quant Biol 36:387–95, 1972.
3 Truan, G, JC Epinat, C Rougeulle, C Cullin, and D Pompon. Cloning and characterization of a yeast cytochrome b5-encoding gene which suppresses ketoconazole hypersensitivity in a NADPH-P-450 reductase-deficient strain. Gene 149:123–7, 1994.
4 Mueser, TC, NG Nossal, and CC Hyde. Structure of bacteriophage T4 RNase H, and 5' to 3' RNA-DNA and DNA-DNA exonuclease with sequence similarity to the RAD2 family of eukaryotic proteins. Cell 85(7):1101–12, 1996.
5 Ceska, TA, JR Sayers, G Stier, and D Suck. A helical arch allowing single-stranded DNA to thread through T5 5'-exonuclease. Nature 382(6586): 90–3, 1996.
| Cn3D 2.0 Feature Summary |
The following is a summary of the capabilities of Cn3D 2.0: Data Input Sequence Alignment Rendering Color Schemes Residue Labeling Animation Operating Systems Cn3D 2.0 may be obtained
from http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/Structure/cn3d.html. |
An online manual for Cn3D is available at http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/Structure/cn3dhelp.html. |
The blastp algorithm finds matches between a protein query sequence and sequences from a database. In such a scheme, hypotheses as to the biological function of the query are not incorporated into the search strategy. Such hypotheses, however, have the potential to focus a similarity search on a small subset of database sequences and thereby draw attention to subtle similarities, with the added benefit of reducing computational loads. Pattern Hit Initiated (PHI) BLAST implements an "hypothesis-driven" search strategy by restricting a BLAST search to those protein sequences within a database that contain a specified pattern, or motif.1
Following the link to PHI-BLAST from NCBI’s main BLAST form leads to a Web form that is similar in layout to the Basic and Advanced BLAST forms but differs from these in that it requires as input both a protein query sequence and a query seed pattern. This seed pattern must occur at least once in the query sequence.
The PHI-BLAST Search Strategy
Each instance of the seed pattern found in a database sequence is first matched with each instance of the pattern found in the query sequence. PHI-BLAST then attempts to construct an optimal local alignment including and extending outward from each seed pattern match. The quality of the resulting alignment is evaluated in a manner similar to that for a Gapped BLAST alignment and is expressed as an expectation value. By coupling a pattern search to a local alignment of measurable significance, PHI-BLAST allows the importance of a motif found in a target sequence to be evaluated within the context of the surrounding sequence. The output resulting from a PHI-BLAST search is presented in the PSI-BLAST (Position-Specific Iterated)2 format and can be used to construct a position-specific profile for use in a subsequent PSI-BLAST search.
Construction of the Seed Pattern
The seed patterns used by PHI-BLAST must be
expected to occur less frequently than once per 5,000 database
residues; any pattern with four fully specified residues
satisfies this constraint. Patterns are constructed using
one-letter amino acid codes and following the PROSITE3 database syntax for the
specification of ambiguity. As an example, the pattern
[LIVMF]GExRVx(5,11)ATx(5)GKS
translates as "Any one of L,I,V,M, or F, GE, any residue,
RV, 5 to 11 ambiguous residues, AT, 5 ambiguous residues,
GKS."
An Example: Apoptosis and Plant Disease Resistance
The utility of PHI-BLAST can be seen in the case of the analysis of the Caenorhabditis elegans protein CED4 (PID 231729), a regulator of programmed cell death, or apoptosis. A Gapped blastp search of the nr (nonredundant) database using the sequence of CED4 as the query yields a statistically significant match to a single protein, Apaf-1 (PID 2961373), a human apoptosis regulator with similarities to plant disease-resistance proteins. Hence a tenuous link between the C. elegans CED4 apoptosis regulator and plant disease-resistance proteins is suggested. This link can be made directly with PHI-BLAST, however, by making use of the observation that the sequence of CED4 contains a P-loop motif characteristic of a variety of ATPases.
The hypothesis that the P-loop motif is of biological significance can be exploited by conducting a PHI-BLAST search using this motif, specified as [GA]xxxxGK[ST], as the query seed pattern. This PHI-BLAST search finds the match to Apaf-1 noted above as well as a second statistically significant match to the CED4 query sequence with an expectation value of 0.035. This new match is to the Arabidopsis thaliana protein T7N9.18 (PID 2213598), a plant disease-resistance protein. Thus, although the link between the C. elegans CED4 protein and plant disease-resistance proteins can be made through Apaf-1 using Gapped BLAST, this link can be made directly using PHI-BLAST.
Stand-alone PHI-BLAST
Stand-alone PHI-BLAST is implemented within the program blastpgp. This program is found in the BLAST archives for Windows and Unix platforms at ftp://ncbi.nlm.nih.gov/blast/executables/.
Notes
1 Zhang, Z, AA Schaffer, W Miller, TL Madden, DJ Lipman, EV Koonin, and SF Altschul. Protein sequence similarity searches using patterns as seeds. Nucleic Acids Res 26, 3986–90, 1998. [Follow the Reference link from the PHI-BLAST page to read this paper online.]
2 Altschul, SF, TL Madden, AA Schaffer, J Zhang, Z Zhang, W Miller, and DJ Lipman. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–402, 1997.
3 Bairoch, A, P Bucher, and K Hofmann. The PROSITE database, its status in 1997. Nucleic Acids Res 25:217–21,1997.
GenBank submissions for sets of multiple sequences are on the rise, accompanied by questions about how to submit them. These submissions include identical, or nearly identical, sequences from a single gene obtained from different sources (e.g., different specimens, isolates, strains, geographic regions) or as part of a population, phylogenetic, or mutation study. When submitting such sequences, prepare a separate GenBank record for each strain, isolate, or individual in the set. The multiple sources should not be documented as part of a single, merged sequence record.
GenBank is an archival database in which each entry constitutes the record of an independent sequencing experiment. If two such experiments result in identical data, then each experimental result is recorded separately in GenBank under a unique accession number. No attempt is made to reduce redundancy by merging sequence records. This system of record-keeping is analogous to the system used by most scientists to document their experiments and ensures that no information is lost.
In studies involving multiple sequence sets, each sequence represents a different experiment, which should be documented in GenBank. It is scientifically useful to know that the described sequence is found to be identical (or nearly so) in different isolates of the same organism or in various populations, and to keep track of the number of times the sequence is represented in a population.
Sequin is the best program for submitting multiple sequence sets, although BankIt can also be used (http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/BankIt/). Sequin allows you to represent a sequence set specifically as a phylogenetic, population, or mutation study. Sequin also facilitates making extensive annotations. Biological features are annotated on a single master record in the sequence set and then automatically propagated to the other members of the set. This propagation of features is possible through the alignment of the sequences. It is, therefore, as simple to use Sequin to prepare hundreds of sequences for submission as it is to prepare a handful. To learn more about Sequin, see http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/Sequin/.
GenBank has been phasing out use of Authorin as a data submission tool for the past 2 years. Currently, less than 1% of all GenBank submissions are made using Authorin. Effective January 1, 1999, Authorin submissions will no longer be accepted.
Submitters are requested to use BankIt on the Web or Sequin, a stand-alone program for PC, Macintosh, and Unix platforms, for both new submissions and database updates. Bulk submissions of EST, STS, and GSS sequences will continue to be handled by customized submission forms available via the NCBI home page. Procedures for HTG submissions will continue to be customized for each high throughput sequencing center.
Using the Genes and Disease Web SiteThe Human Genome Project is now on target to map and sequence the whole of the human genome by 2003. When complete, it will serve as a blueprint for the molecular biology of human life. GeneMap ’98, described on page 1 of this issue, is a forerunner to this complete sequence map; it defines the locations of 30,000 clusters of identical ESTs (small pieces of expressed genes). Each cluster represents a human gene, and so about half of all human genes are on GeneMap ’98. Many human diseases are known to be caused by mutations in one or more genes. As an accompaniment to GeneMap ’98, the Genes and Disease Web site (available as a link from GeneMap '98 to http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/disease/) provides overviews of about 60 human diseases for which the involvement of a specific gene(s) is indicated. The diseases on the Genes and Disease page are grouped by chromosomal location as well as by their biochemical and physiological characteristics. Genetic disease categories include cancer, nervous system, and metabolism, as well as several others. Following the Metabolism link, for example, leads to a synopsis of metabolic disorders including links to nine specific metabolic disorders. A click on the second disease link, Atherosclerosis, produces a description of the symptoms and what is known of the genetic origins of the disease. For example, a protein called apolipoprotein E, whose gene resides on chromosome 19, is implicated in atherosclerosis. To the left of the entry for atherosclerosis are links to other resources, including GeneMap ’98, PubMed, sequences in GenBank, and OMIM. These links provide access, respectively, to the human chromosomal map in the environs of the apolipoprotein E gene, literature references to apolipoprotein E, the GenBank sequence record for apolipoprotein E, and the OMIM entry for atherosclerosis. By using the links to individual chromosomes provided at the top of the page, it is possible to survey human genetic disease by chromosome. The link to chromosome 1 provides a graphical representation of chromosomes 1 to 4. The chromosomes are drawn to scale and include the G-bands familiar to cytologists and geneticists. Disease loci are labeled as hyperlinks positioned so as to indicate the map location associated with the disease-causing gene. Following one of these hyperlinks leads to the page describing the disease. |
The Advanced BLAST service available on the Web now allows greater flexibility in the use of the BLAST server. Using a new input box, it is possible to limit a BLAST search to sequences belonging to a particular organism or taxonomic group by typing in a genus and species or a group name. An accompanying list box provides a selection of prominent organisms from which to choose. The new page also offers a choice between the default BLOSUM62 comparison matrix, two additional BLOSUM comparison matrices, and two PAM comparison matrices. A check box now provides a means to toggle between gapped and ungapped alignments. Blastx users will find a list box allowing a selection from 13 genetic codes useful.
BLAST output options have also been extended. A list box now provides a selection of four multiple alignment schemes, called master-slave alignments, in addition to the traditional pairwise BLAST alignment format, which is still the default. Try out the new Web BLAST via the Advanced BLAST link on NCBI’s BLAST Web page at http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/BLAST/.
Single nucleotide polymorphisms (SNPs) are the most common variations in the human genome, occurring once every 100 to 300 base pairs. Because of the volume of data represented by SNPs, a database of SNPs is expected to greatly facilitate large-scale associative genetics studies concerned with the linkage between sequence variation and heritable phenotypes. In collaboration with the NIH National Human Genome Research Institute, NCBI has established dbSNP as a central, public, repository for SNP data as well as for data on short insertions or deletions. The database includes information on sequence variations within individuals and populations as well as descriptions of the assay conditions used to detect each variant. Systems for the integration of dbSNP with other genomic data at NCBI, including GenBank, are also under development.
dbSNP is accessible under "What’s New" on the NCBI home page. SNP sequence submissions are now being accepted via e-mail to snp-sub@ncbi.nlm.nih.gov. Information on how to submit SNP data is available from the dbSNP home page.
GenBank broke the two billion base pair threshold with the October release of GenBank 109.0, just 14 months after hitting the one billion base pair mark in August 1997. These billions of bases are contained in 2.8 million GenBank records.
This GenBank milestone came just in time for the 10th anniversary of NCBI. Established by Public Law 100-607 on November 4, 1988, NCBI was charged with a mission to create and facilitate use of automated systems for storing and analyzing molecular biology data and to conduct research in computational molecular biology. It assumed responsibility for GenBank in 1992.
In the database development arena, NCBI set out to provide integrated access to molecular biology information, beginning with the important step of incorporating translated protein sequences into GenBank as an integral part of the DNA sequence record. Parallel goals were to develop effective sequence analysis tools and to organize and consolidate data in ways that facilitate research. Development of the Entrez retrieval system provided a flexible and powerful platform for expanding integrated access to 3D structures, genome mapping data, a phylogenetic taxonomy, and the published literature. The BLAST sequence analysis programs have undergone continued development since the original algorithm was published in 1990 and are used extensively worldwide. Databases such as UniGene, GeneMap ’98, and Clusters of Orthologous Groups are the result of significant research on data organization, consolidation, and analysis. They provide meaningful views of the underlying data and enhance the usefulness of data resources to the research community.
In the research arena, NCBI has assembled a multidisciplinary group of intramural investigators to conduct research in computational molecular biology and text retrieval. In addition to their contributions to basic science, these investigators serve as a wellspring of new methods for applied research activities. Areas of concentration include gene organization and genome analysis, biomolecular structure modeling and prediction, theory of sequence analysis, and statistical approaches to text retrieval.
In the coming years, NCBI looks forward to expanding its genomic data resources as the pace of discovery continues to increase.
Altschul, SF. Generalized affine gap costs for protein sequence alignment. Proteins 32(1):88–96, 1998.
Aravind, L, DD Leipe, and EV Koonin. Toprim-a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res 26(18):4205–13, 1998.
Baxevanis, A, and BFF Ouellette, eds. Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins. New York: John Wiley & Sons, 1998.
Ermolaeva, O, M Rastogi, KD Pruitt, GD Schuler, ML Bittner, Y Chen, R Simon, P Meltzer, JM Trent, and MS Boguski. Data management and analysis for gene expression arrays. Nat Genet 20(1):19–23, 1998.
Galperin, MY, DR Walker, and EV Koonin. Analogous enzymes: independent inventions in enzyme evolution. Genome Res 8(8):779–90, 1998.
Kulaeva, OI, EV Koonin, JC Wootton, AS Levine, and R Woodgate. Unusual insertion element polymorphisms in the promoter and terminator regions of the mucAB-like genes of R471a and R446b. Mutat Res 397(2):247–62, 1998.
Makalowski, W, and MS Boguski. Evolutionary parameters of the transcribed mammalian genome: an analysis of 2,820 orthologous rodent and human sequences. Proc Natl Acad Sci USA 95(16):9407–12, 1998.
Wilbur, WJ. Accurate Monte Carlo estimation of very small P-values in Markov chains. Comput Stat 13:153–68, 1998.
Yedavalli, VR, C Chappey, and N Ahmad. Maintenance of an intact human immunodeficiency virus type 1 vpr gene following mother-to-infant transmission. J Virol 72(8):6937–43, 1998.
Zhang, Z, AA Schaffer, W Miller, TL Madden, DJ Lipman, EV Koonin, and SF Altschul. Protein sequence similarity searches using patterns as seeds. Nucleic Acids Res 26(17):3986–90, 1998.
NCBI News is distributed three times a year. We welcome communication from users of NCBI databases and software and invite suggestions for articles in future issues. Send correspondence to NCBI News at the address below.
NCBI News
National Library of Medicine
Bldg. 38A, Room 8N-803
8600 Rockville Pike
Bethesda, MD 20894
Phone: (301) 496-2475
Fax: (301) 480-9241
E-mail: info@ncbi.nlm.nih.gov
Editors
Dennis Benson
Barbara Rapp
Writer
David Wheeler
Managing Editor
Roseanne Price
Graphics and Production
Veronica Johnson
Design Consultant
Troy M. Hill
In 1988, Congress established the National Center for Biotechnology Information as part of the National Library of Medicine; its charge is to create information systems for molecular biology and genetics data and perform research in computational molecular biology.
The contents of this newsletter may be reprinted without permission. The mention of trade names, commercial products, or organizations does not imply endorsement by NCBI, NIH, or the U.S. Government.
NIH Publication No. 99-3272
ISSN 1060-8788
ISSN 1098-8408 (Online Version)