NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Frink RJ. Inflammatory Atherosclerosis: Characteristics of the Injurious Agent. Sacramento (CA): Heart Research Foundation; 2002.
“ It seems certain that many patients do survive plaque fissuring without developing any symptoms…”
MJ Davies, et al., [120]
Chronicity of UPs
Nagatomo [144] showed UPs persisting as chronic ulcerations for weeks, months, or years before progressing to luminal stenosis, thrombosis, and acute events. Other investigators [145–147] showed complex lesions, present in patients with unstable angina, persisting as chronic lesions, and progressing to stenosis and acute coronary events. Our results, Table 4, show UPs were ubiquitous, multiple, occurring in the same artery, but at a different location, or in a different artery from the one containing the culprit thrombus. These ulcerations did not develop simultaneously, and are of different ages [57,148]. If UPs are of a different age, can their age be determined? If so, what is their natural history? Do they reseal and progress, forming a larger plaque with a larger necrotic core, as Davies suggests [120,149,150], or do they persist as chronic ulcerations (57,148) without resealing and without growth of the necrotic core? Nakagawa [151] noted that PUs associated with an occlusive thrombus were still present one month after thrombolysis, but the angiographic degree of occlusion had decreased. Plaques do ulcerate, empty, and decompress with regression of stenosis, but they may not quickly resolve and reendothelialize. The UPs shown in Figure 22, Figures 22A and 22B, show no evidence of resolution, no platelet aggregation, mural thrombosis, recanalized thrombus or other evidence of repair. They do not present a histologic picture of actively resolving injury.
We hypothesize that UPs, once the fibrous cap has been breached, will not permanently reseal, and the atheroma will not completely resolve until the necrotic core is completely empty. Without complete drainage of the necrotic core the factors that caused the plaque to ulcerate in the first place are still present including the active, destructive IA, the toxic core contents, and the various proteases. We further hypothesize that many UPs without thrombosis are, in reality, indolent, festering, inflammatory, chronically draining lesions that persist indefinitely until either fibrotic stenosis without associated atheroma or occlusive thrombosis develops.
Depth and Extent of UPs
The relationship between luminal stenosis and the depth and extent of the 109 UPs without thrombosis, shown in Table 4, is presented in Table 5. The UPs were graded in the following manner to determine the extent, depth, and severity of the ulceration [57]: A Grade I ulceration involved only the inner surface of the intima, and was confined to a single coronary segment. A Grade II ulceration was also confined to a single segment, but penetrated to the depths of the intima. A Grade III ulceration involved two adjacent segments, and a Grade IV involved three or more contiguous segments. Deep and extensive UPs were as frequent in plaques with <50% stenosis as they are in plaques with 70 to 79% stenosis. The frequency, depth, and extent of the UPs was not related to the severity of luminal stenosis. Severe luminal stenosis is not required for PU, and severe luminal stenosis does not necessarily develop before PU. These findings support other investigations that show PU is not related to plaque size or the degree of luminal stenosis [152,153].
Table 6 compares the frequency of atheromas, adventitial inflammation, and calcification at the site of all UPs associated with or without luminal thrombosis in the 83 patients presented in Tables 4 and 5. Virtually all UPs are associated with inflammation, calcification, and a necrotic core, and all are objective signs of active atherosclerotic disease (See Chapters 3,4,5) [57]. We conclude inflammation and calcification play some role in ulceration of the necrotic core (53,63).
UPs and Fibrotic Luminal Stenosis
If UPs without thrombosis persist indefinitely as chronic lesions, what is the relationship between chronic UPs and the development of fibrotic, non-atheromatous, luminal stenosis? Chronic inflammatory diseases, in general, are characterized by inflammation, tissue destruction, and a FP response at the site of the inflammation [47]. We postulate that the bodily response to chronic inflammation in the coronary artery will be the same or very similar to the bodily response to chronic inflammation elsewhere in the body, i.e., fibrous tissue will form around the lesion. We would expect an FP response at the site of a chronic, persistent UP to be manifested by the development of progressive, fibrotic, luminal stenosis. Chronic UPs and the associated release of growth factors could account for rapid progression of insignificant lesions [154]. Luminal stenosis, in this circumstance, would be directly related to the inflammatory activity surrounding the chronic UP. It would be the result of and would develop after, not before, the plaque ulcerates. The luminal stenosis would then increase, not because the plaque reseals but because the UP stimulates a FP response similar to healing by secondary intention [57,148,150]. The UP does not resolve, but persists as luminal stenosis increases. The longer the UP persists, the greater will be the fibrotic luminal stenosis. More specifically, the severity of fibrotic luminal stenosis found in association with an UP may reflect the age of the UP.
To continue this hypothesis, the 28 UPs without thrombosis in the 70–79% stenosis range (Table 5), have been present longest and have resulted in the most severe stenosis. The 27 UPs associated with <50% stenosis are the most recently formed, with the least fibrotic stenosis. The age of the 18 UPs associated with 50–59% stenosis and the 24 associated with the 60–69% stenosis would be intermediate in age. These UPs without thrombosis may then persist as chronic UPs until the underlying stenosis approaches 80% of the cross sectional area, at which point the conditions and substrate favoring thrombosis are present.
Sudden development of acute coronary syndromes as a result of thrombotic occlusion naturally leads to the assumption that the UP beneath the acute thrombus is also an acute event. This assumption is unproved. We know of no way in which the age of an UP can be accurately determined histologically. The chronic UP and the chronic FP response may have been actively brewing for months or years without overt symptoms, before the acute event.
Component versus Complication
The well known association between UPs, thrombus formation, and acute coronary events does not show PU to be a complication of the disease process, but only shows that it has the potential to develop into a complication under certain circumstances. A complication, strictly speaking, describes a secondary disease that aggravates a previous one. Since there are many UPs not associated with thrombosis or other pathologic sequelae, and without apparent clinical effects [63,120], PU, per se, cannot technically be called a complication. If PU is a component of active disease, it is not necessarily a pathologic event that must be, should be, or even can be prevented [155].
The distinction between PU as a component rather than a complication gives us insight into the nature of the IA and the disease process. It also alters our approach to the prevention and treatment of the disease. If PU is a component of active atherosclerotic disease, then all inflammatory atheromas may be expected to ulcerate at some point, the large vulnerable plaques [150,153] posing the greatest danger. If such vulnerable plaques could be identified in vivo, before ulceration, it may be possible to devise an intervention to control or remove the plaque before ulceration develops, or devise a treatment to neutralize the effects of ulceration when it does occur.
If PU is a complication of active disease, then it is a random event, possibly affecting only a few plaques, with many atheromas never undergoing ulceration. Identifying vulnerable plaques and performing an intervention, may not be appropriate because not all such plaques may require intervention. Devising treatments to manage plaques that will spontaneously ulcerate and drain at some point in the future is decidedly different than devising treatments for complications that may or may not develop.
Early PU Is the Norm
We believe that the norm is for plaques to ulcerate early in their development. It is their failure to do so that is abnormal and pathologic in nature. Failure of a plaque to ulcerate early leads to the formation of a large necrotic core, such as illustrated in Figure 6, and to the large vulnerable plaque. Early ulceration of a necrotic core may be quite beneficial in the sense that it is an efficient method of reverse transport, debulking and decompressing the plaque core by extruding plaque contents [15,57].
Absence of Thrombus
Table 4 shows that, consistent with previous investigations [57,120,156,157], many UPs within the coronary tree of patients who have died of ACD are not associated with luminal thrombosis. The observations in this study of 83 patients demonstrate that PU is not synonymous with thrombosis, and thrombosis does not automatically form on exposed subendothelial tissue [158]. Exposed subendothelial tissue, including collagen, may not be as thrombogenic as previously believed [159].
Figure 22 shows several examples of UPs without associated thrombosis. They appear to be open, draining, abscess-like cavities. The fibrous cap has been breached, exposing plaque contents and subendothelial collagen to flowing blood, with resulting extrusion and partial emptying of the necrotic core (Figures 22A-C). Why is thrombus absent if plaque contents or exposed subendothelial tissue are so thrombogenic [160]?
The absence of thrombus in some UPs may be explained by the presence of MMPs and other proteases within atheromas. These enzymes dissolve tissue proteins, play an important role in degrading and digesting dead or damaged tissue within the necrotic core, and erode the surrounding fibrous tissue, including the fibrous cap [114,161]. T cells trigger macrophages to secrete MMPs, showing these MMPs also to be a component of active, atherosclerotic disease [53,162,163]. MMPs are also capable of digesting fibrin and of preventing thrombus formation and have also been shown to negatively affect platelet adhesion and aggregation [163]. Thus, the pro-teases contained within atheromas may prevent thrombus formation in UPs, particularly those associated with insignificant luminal stenosis, by contributing to inherent thrombolysis.
Figures 22D, E illustrates a large UP with a thrombus formed on a remnant of a fibrous cap, with no evidence of thrombus on the exposed surface at the base of the core. Most plaque contents are gone from this plaque with only residual degenerative tissue lining the margins of the core. In this situation, a thrombus has formed on one portion of an UP, but not on other exposed areas of the extensive ulceration, revealing a difference in the thrombogenicity of these two adjacent tissues [160]. If the tissue around the periphery of the necrotic core is markedly thrombogenic, we would expect histologic evidence of platelet aggregation and/or mural thrombosis in the area. The difference in thrombogenicity may be related to the MMPs and other enzymes that continue to be present at the margins of the necrotic core, even after the core contents have been removed [63,114].
In Review
PU is a natural component, not a complication, of active progressive atherosclerotic disease, often occurring early in plaque development. PU is related to the activity of the IA, and it results in the debulking and decompression of atheromas. It is an efficient form of reverse transport. The UP without thrombosis provides insight into the nature of the IA and to the pathogenesis of PU, fundamental to our understanding of active atherosclerotic disease. UPs may persist as chronic festering, inflammatory lesions for indefinite periods, giving rise to fibrotic luminal stenosis and ultimately to occlusive thrombosis.
Figures
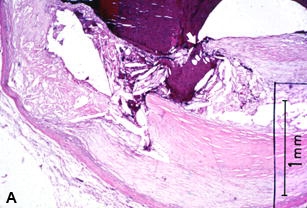
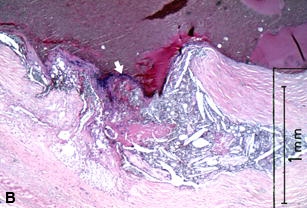
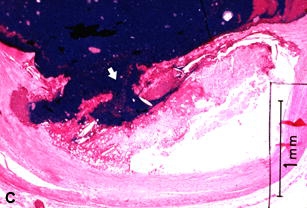
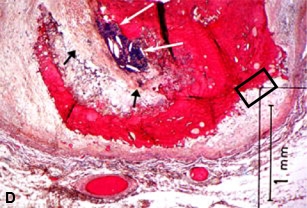
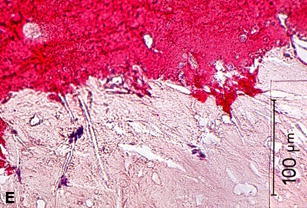
Figure 22
A–C are examples of UPs without luminal thrombosis and without significant luminal stenosis in 3 different patients. The fibrous cap has been breached (fat white arrows) in all patients, portions of the necrotic core have been extruded and replaced by injection mass. Inflammatory cells are scattered in the adventitia (not shown).These UPs were not considered to be the culprit lesions. A, Mid-LAD artery of a 53-year-old white female who died SCD outside the hospital, 6 weeks following an acute subendocardial infarction. B, Main left coronary artery of a 69-year-old white female who died in hospital of cardiogenic shock associated with an AMI. C, Proximal CIRC artery of a 67-year- old white male who died of cardiogenic shock following an AMI. All stains H & E. D, Section taken from the mid-LAD artery just proximal to an occluding thrombus of a 43-year-old white male who died SCD out of hospital. A portion of the fibrous cap (short black arrows) is involved with thrombus (long white arrows), primarily on the luminal surface. Most of the core contents have been extruded. E, High-power view of rectangle in D, showing no thrombi have formed on the exposed surface. D & E = PTAH stain.
Tables
Table 4Comparison of the incidence of coronary thrombosis and of ulcerated plaques without thrombosis with the degree of luminal stenosis in four different acute coronary syndromes
| Degree of Luminal Stenosis (%) | Totals | (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| <50 | 50–59 | 60–69 | 70–79 | 80–89 | 90–99 | 100 | ||||
| Cardiogenic Shock | UP | 5 | 11 | 7 | 10 | 2 | 3 | 38 | 48 | |
| No. 28 | Tc | 2 | 4 | 6 | 8 | 21 | 41* | 52 | ||
| Total | 5 | 11 | 9 | 14 | 8 | 11 | 21 | 79 | ||
| SCD w/ AMI | UP | 3 | 4 | 6 | 4 | 1 | 1 | 19 | 44 | |
| No. 18 | Tc | 2 | 2 | 8 | 12 | 24* | 56 | |||
| Total | 3 | 4 | 6 | 6 | 3 | 9 | 12 | 43 | ||
| SCD w/o AMI | UP | 15 | 3 | 8 | 7 | 3 | 36 | 61 | ||
| No. 26 | Tc | 4 | 4 | 6 | 9 | 23* | 39 | |||
| Total | 15 | 3 | 8 | 11 | 7 | 6 | 9 | 59 | ||
| Cardiac Rupture | UP | 4 | 3 | 7 | 1 | 1 | 16 | 53 | ||
| No. 11 | Tc | 3 | 2 | 3 | 6 | 14* | 47 | |||
| Total | 4 | 6 | 7 | 3 | 4 | 6 | 30 | |||
| Totals | UP | 27 | 18 | 24 | 28 | 7 | 5 | 109 | 52 | |
| No. 83 | Tc | 5 | 10 | 14 | 25 | 48 | 102* | 48 | ||
| Total | 27 | 18 | 29 | 38 | 21 | 30 | 48 | 211 | ||
UP = Ulcerated plaque without thrombosis; Tc = Occlusive coronary thrombus; SCD = Sudden cardiac death; AMI = Acute myocardial infarction; * = p =<0.001 (Reprinted from reference 57 with permission)
Table 5Comparison of the degree of luminal stenosis with the severity of the plaque ulceration in 109 ulcerated plaques without thrombosis
| Severity of UP | Degree of Luminal Stenosis (%) | Totals | |||||
|---|---|---|---|---|---|---|---|
| (Grade) | <50 | 50–59 | 60–69 | 70–79 | 80–89 | 90–99 | |
| I | 7 | 6 | 4 | 6 | 1 | 1 | 25 |
| II | 9 | 5 | 4 | 7 | 1 | 3 | 29 |
| III | 5 | 2 | 6 | 6 | 1 | 20 | |
| IV | 6 | 5 | 10 | 9 | 4 | 1 | 35 |
| Total | 27 | 18 | 24 | 28 | 7 | 5 | 109 |
UP = Ulcerated plaque without thrombosis. (Reprinted from reference 57 with permission)
Table 6Comparison of the incidence of inflammatory cell infiltrates, calcification, and necrotic plaques at the site of ulcerated plaques without thrombosis and with occlusive thrombotic lesions. The incidence of these three lesions in the entire group and in the control patients is also provided for comparison
| CATEGORY | # of lesions | IC | % | CA | % | NP | % | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Ulcerated Plaques | ||||||||||
| < 50% Stenosis | 27 | 26 | 96 | 26 | 96 | 25 | 93 | |||
| > 50% Stenosis | 82 | 78 | 95 | 70 | 85 | 82* | 100 | |||
| Total UP | 109 | 104 | 95 | 96 | 88 | 107 | 98 | |||
| Thrombotic Lesions | 102 | 93 | 91 | 95 | 93 | 94 | 92 | |||
| Entire Group | # of Sections | IC | % | CA | % | NP | % | |||
| 83 patients | 7036 | 3597 | 51 | 2654 | 38 | 2195 | 31 | |||
| 22 controls | 1789 | 295 | 16 | 221 | 12 | 101 | 6 | |||
IC = Inflammatory cell infiltrates; CA = Calcification; NP = Necrotic plaque; UP = Ulcerated plaque without thrombosis; * = (p = <0.02) (Reprinted from reference 57 with permission)