

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Abbreviations
RT-PCR: reverse transcription polymerase chain reaction
TRAP: telomeric repeat amplification protocol
hTERT: human telomerase reverse transcriptase
hTERC: human telomerase RNA component
hTEP1: human telomerase associated protein 1
DC: dendritic cell
ALT: alternative lengthening of telomeres
FISH: fluorescence in situ hybridization
SFT: solitary fibrous tumor
SV40 Ltag: simian virus large T antigen
M1/2: mortality stage 1/2
PRINS: primed in situ
CGH: comparative genomic hybridization
LOH: loss of heterozygosity
TSG: tumor suppressor gene
Introduction
Malignant mesothelioma (MM) is an aggressive malignancy of the mesothelial cell (MC) lining the body cavities. In western Europe, its incidence is expected to rise till 2020 because of the widespread use of asbestos fibers, the causative agent, and the decade-long interval between exposure and symptomatology. Very often, pathologists have great difficulties in discriminating mesothelioma from benign mesothelial lesions. In particular, the differential diagnosis between epithelial-type mesothelioma and hyperplastic mesothelium, as well as between the sarcomatoid type mesothelioma and fibrous chronic pleural inflammation, really is a challenge. A tally of techniques have been developed to solve the problem, but none suffices today. On the other hand, oncologists face the treatment-refractory character of this cancer. Despite the advent of new strategies like photodynamic therapy, immunotherapy and gene therapy, prognosis remains poor with a median survival ranging from 12 to 17 months and a 5-year survival of less than 5%. Interestingly, the disease becomes symptomatic decades after exposure to carcinogenic asbestos fibers, suggesting the long-term survival (immortalization) of premalignant cell clones.
The process of immortalisation is linked to telomere biology. Gradual erosion of telomeres, the very ends of human chromosomes, eventually limits the replicative life span of somatic cells and this process can be regarded as an ultimate tumor suppressor mechanism, eliminating cells that have accumulated genetic alterations. The enzyme telomerase elongates telomeres and thus prevents chromosome erosion and telomerase was found to be activated in over 85% of human cancers.
Telomerase has been welcomed by both diagnosis-oriented pathologists and treatment-oriented clinicians. Since telomerase seems closely associated with malignancy, telomerase is hoped to be an early indicator of cancer and a new therapeutic target common to most cancers. In section 2, we report our results of semiquantitative measurements of telomerase activity in extracts from 22 primary pleural mesotheliomas, four mesothelioma cell lines, and six short-term mesothelial cell cultures from normal pleura. Twenty of the 22 primary mesotheliomas (91%) and all tumour-derived mesothelioma cell lines were telomerase positive. Different levels of enzyme activity were observed in the tumours of different histological subtypes. Telomerase activity could not be detected in the six normal mesothelial cell cultures nor in two mesotheliomas. In section 3, we determined the expression of hTERC, hTEP1, and hTERT, in 16 pleural MMs and 4 MM-derived cell lines and in 6 MCCs. RT-PCR analysis revealed that hTERT mRNA expression parallels the activity status documented by the TRAP assay, whereas hTERC and hTEP1 mRNA are commonly expressed in all cancerous and noncancerous serosal cells and tissues. The TRAP and RT-PCR analyses indicate that hTERT expression is rate limiting for human telomerase activity and that reactivation, rather than up-regulation, of hTERT expression can play a critical role in MM carcinogenesis. As detailed in section 4, both the TRAP and the RT-PCR assays are hampered by the loss of tissue morphology and the need for fresh or frozen tissue samples, which is disadvantageous when studying small, archived reactive or hyperplastic lesions. We report our attempts and summarize the efforts of others to detect the hTERT protein at the cellular level using the first generation of commercially available anti-hTERT antibodies. Unfortunately, the antibodies were not found reliable and experiments with newer antibodies are ongoing. In section 5, we describe preliminary in vitro experiments in which we were able to stop the growth of 2 out of 3 telomerase-positive, MM-derived cell lines, using antisense oligonucleotides.
Telomerase Activity in Malignant Mesothelioma
In 1994, the TRAP assay was published, allowing biochemical detection of telomerase activity in fresh or frozen cell or tissue lysates.1 In this assay, an aliquot of the test lysate is incubated with a mixture containing a synthetic telomere-like oligonucleotide, which, in the presence of telomerase activity, will be elongated by a number of hexameric repeats. In a conventional polymerase chain reaction, the mixture of telomerase products, which differ in size by a multiplicand of 6 bases, is amplified and separated on polyacrylamide gels. In case of telomerase activity, a typical 6-bp incremental ladder is visualized. Numerous modifications of the original protocol have been made in order to improve reliability, linearity and sensitivity, to exclude false-positive results due to primer-dimer formation, to quantify enzyme levels, and to exclude false negativity due to the presence of Taq polymerase inhibitors.25 We perform quick, safe and effective detection of nonlabeled amplicons using the DNA stain ethidium bromide, and a CCD camera-coupled software package (Gel Doc 1000 Molecular Analyst, BioRad Laboratories GmbH, Germany) (Fig. 1).

We measured telomerase activity in 22 primary pleural mesotheliomas, 2 benign solitary fibrous tumors of the pleura, 4 mesothelioma cell lines, and 6 short-term mesothelial cell cultures using the aforementioned nonisotopic TRAP assay.6 We analyzed telomerase activity in a highly proliferative mesothelial cell culture as well as in mesothelial cells that showed features of (pre)senescence (Fig. 2). Telomerase activity was detected in 20/22 (91%) of primary mesotheliomas, and all tumor-derived mesothelioma cell lines (Fig. 3). Levels of enzyme activity were semiquantitatively graded using lysate dilution series, and different levels were observed in the tumors of different histological subtypes (see Table 1). Telomerase activity could not be detected in the 6 normal mesothelial cell cultures nor in 2 mesotheliomas. Unexpectedly, both benign solitary fibrous tumors showed strong telomerase activity.
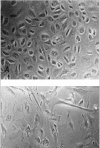

Table 1
Telomerase activity profiles of individual mesotheliomas and solitary fibrous tumors in relation to histological subtype. (Reproduced from ref , with permission from the publisher).
The finding of telomerase activity in over 90% of mesotheliomas is in agreement with the reported activity in a variety of other cancer types and is consistent with the hypothesis that telomerase activation may be a feature of carcinogenesis in mesothelioma as perhaps in many other cancers. Carcinogenesis is recognized as a multistep process resulting from the accumulation of sequential genetic alterations in a cell. The very long latency period that is characteristic of mesothelioma suggests that multiple cumulative genetic, cytotoxic and proliferative events occur during the tumorigenic conversion of the progenitor cell. Of the known changes that we and others investigated in mesothelioma, the most frequent are in the p16 and NF2 genes (for a synopsis, see ref. 7). According to the telomere-senescence model, such genetically altered premalignant cells finally may be eliminated before they ever can develop a full-blown malignant phenotype. Conversely, the presence of telomerase activity in mesothelioma may indicate that these cells possibly can overcome the restraint of their finite lifespan and are no longer hampered in their clonal evolution towards a more malignant phenotype. We did not detect telomerase activity in normal mesothelial cells. There is evidence that mesothelioma originates from surface mesothelial cells rather than from a ‘multipotential’ subserosal stem cell, although this has not been established beyond all doubt.8 Assuming the former scenario is correct, the absence of detectable telomerase activity in mesothelial cells, in contrast to the presence of activity in mesotheliomas, could fit with the interpretation that most adult tumors develop from telomerase-negative precursors after telomerase reactivation.9 In contrast, others may interpret these results as evidence for the abovementioned stem cell origin of mesothelioma, arguing that these telomerase-positive tumors indeed arise from telomerase-positive stem cells without the need for a reactivation step.10 Importantly, we neither detected telomerase in proliferating nor in resting mesothelial cells. This result conceivally argues against a model in which telomerase is regarded merely as a proliferation marker.11 Indeed, extensive TRAP analyses and hTERC in situ hybridization studies showed a similar (but not identical!) distribution pattern of telomerase activity and/or hTERC RNA and the Ki67 (Mib-1) proliferation marker.12,13 Other cell types, that scored telomerase negative in vivo, appeared competent to express telomerase when subjected to a sufficient proliferative stimulus in vitro.14 Therefore, it is currently only valid to apply the classic reactivation model to a particular organ system when the cell of origin is effectively telomerase negative, not just in its in vivo state but also when subject to ‘excessive’ growth stimulation.15 This controversial issue was subject of a more detailed study, in which we determined telomerase activity and hTERT mRNA expression in primary melanocyte cultures.16 Here too, highly proliferating melanocyte cultures, showing nuclear staining for Ki67 and for KiS2 in 65% and in 35% of the cells, respectively, lacked telomerase activity and hTERT transcripts. KiS2 binds to an epitope that is present during the entire cell cycle, with exception of the rate-limiting G1 phase, thereby being a more accurate marker of the actively proliferating cell fraction than Ki67.17 The detected strong telomerase activity in both benign solitary fibrous tumors could reflect their hypothesized origin from telomerase-positive, CD34-positive fibroblastic stem cells or their unpredictable clinical behavior. Similarly, Umbricht et al found 19% of benign follicular tumors of the thyroid with detectable telomerase activity and argued that some histologically benign lesions may be precursor lesions of follicular carcinomas.18 Our observation can also indicate that the application of telomerase activity as a marker for malignancy of serosal lesions needs further validation. It is obvious that larger series need to be studied, which should also include unequivocal benign, mesothelium-derived tumors, e.g., adenomatoid tumors. The ‘classic model’ states that telomerase up-regulation is forced by critical telomere erosion beyond the point where cell multiplication normally stops. An in vitro situation is seen during continuous culture of SV40 large T antigen-transformed, telomerase-negative human cells, which eventually undergo ‘crisis’, the condition in which cellular chromosomes are characterized by ultrashort telomeres and that coincides with telomerase activation.19 At present it is not known whether SV40-like viruses, for which DNA sequences have been found by us and others in some mesotheliomas, are responsible for an in vivo equivalent of crisis during mesothelioma carcinogenesis20 (for a review, see ref. 21). The ‘co-selection hypothesis’ states that telomerase is indirectly reactivated as part of a ‘package’ of changes in gene expression that occurs after some other genetic event.22 It remains speculative whether asbestos-induced DNA damage represents such a genetic event. Similarly, others have discussed that telomerase activation is a mere side effect of dedifferentiation.23 We observed different levels of enzyme activity in the tumors of varying grades of differentiation. Because of the limited study size, we did not perform statistical analysis to find any correlation between the level of telomerase activity in the tissue lysates and histologic subtype. Anyway, statistical results may be inaccurate because of weaknesses in the TRAP assay. Indeed, heterogeneity of tumor differentiation within a tissue section as well as morphology is lost during the in vitro biochemical assay. Further, the TRAP assay requires access to fresh or frozen samples, indicating the need for alternatives that can detect telomerase activity at the cellular level in archived materials. Such alternatives will be extremely helpful to study the telomerase status in archived samples of serosal precursor lesions.
Gene Expression Profile of Components of Telomerase in Mesothelioma
TRAPbased activity studies of lysates of normalserosal lining and of preneoplastic mesothelial lesions are hampered by the difficulty of morphological feedback and the impracticability of using fixed/embedded materials, furthering the ongoing search for surrogate markers that could detect telomerase activity at the cellular level. In contrast to hTERC and hTEP1, the discovery of the hTERT gene, was welcomed with great enthusiasm because a strong correlation was found between hTERT mRNA expression and telomerase activity levels in primary cancers and tumor-derived cell lines.24,25 However, conflicting results exist with regard to the expression profile of hTERT in some normal tissues and primary cell lines, questioning whether the differences in hTERT expression in tumor versus normal cells are relative or absolute.26 Moreover, reverse transcription-polymerase chain reaction (RT-PCR)-based expression profiles showed tissue-specific, alternative splicing-based expression in various tumors, cell lines and in normal tissues, suggesting that alternative splicing of hTERT transcripts could control the activity of telomerase in a tissue-specific way.27 We have studied the expression pattern of each telomerase subunit in all mesothelioma tumors and tumor-derived cells as well as in mesothelial cell cultures.28 To detect hTERT mRNA splice variants, primers that span 2 common splicing sites (amplicon size 457 bp), were used, as designed by Ulaner et al27 (Fig. 4).
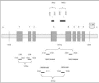
RT-PCR analysis revealed that hTERT mRNA expression parallels the activity level documented by the TRAP assay, whereas hTERC and hTEP1 mRNA are commonly expressed in all cancerous and noncancerous serosal cells and tissues. In accordance with recent studies, our findings indicate that expression of hTERT is rate limiting in the mesothelial organ system, too. Our results further show that the differences in hTERT expression in mesothelioma cells and tissues versus mesothelial cells are absolute and not relative, as cited by Ramakrishnan et al for other cell types.26 This group detected low hTERT expression in renal epithelial cells, prostate epithelial cells and WI38 lung fibroblasts. The absolute absence of hTERT mRNA in mesothelial cells was additionally confirmed using 2 other sets of primers (amplicon sizes of 145 and 281 bp) (Fig. 5). In accordance with the TRAP assay results, we did not see any change in expression between proliferating and resting mesothelial cell cultures, corroborating our arguments against a role for telomerase as just another proliferation marker [data not shown].11 This RNA profiling suggests that hTERT expression, by unclear mechanisms, is reactivated rather than up-regulated during mesothelioma carcinogenesis. Recent cloning and sequence analysis of the hTERT gene promoter revealed the presence of binding sites for transcription factors including the c-Myc proto-oncoprotein.29,30 The latter activates telomerase by inducing expression of its catalytic subunit, indicating that hTERT is a target of c-Myc activity.31,32 Interestingly, we previously found statistically significant higher immunoreactivity for the c-Myc protein in neoplastic compared to nonneoplastic mesothelium.33 Also, recent deletion analysis indicated 2 regions on chromosome 3 (3p21.3p22 and 3p1221.1), where telomerase repressor genes may be located.34 Cytogenetic studies and LOH analyses in a series of 25 mesotheliomas, identified 3p21 as the critically deleted segment with the highest frequencies of allelic loss at the 3p21.3 and at the 3p21.1 locus.35 Using a primer pair that spans 2 splicing sites (a and b), 3 alternatively spliced hTERT transcripts were detected in all telomerase-positive samples, whereas neither the full-length transcript nor any variant could be detected in the mesothelial cells. From hTERT expression studies during human development, it was inferred that alternative splicing of hTERT mRNA might occur in a tissue-specific manner.27 In all mesothelioma tumors and cells, the b deletion variant accounted for a significant fraction of the total hTERT transcripts, with the a-deleted and the a/b-deleted forms being weakly expressed [data not shown]. The significance of the distribution pattern of spliced products awaits knowledge on the role of the individual mRNA variants, and elucidation whether these variants are translated into biologically functional proteins.

Detection of Telomerase at the Cellular Level
From our previous results, we concluded that hTERT expression is rate limiting for telomerase activity, and that hTERT reactivation may play a critical role in mesothelioma carcinogenesis. It still needs elucidation at what stage telomerase is reactivated, and whether precursor lesions express telomerase. Because tissue morphology is lost, both the TRAP assay and RT-PCR are not practical in answering this question. In the international research community, a race is currently ongoing to develop suitable anti-hTERT antibodies. We tested a commercially available anti-hTERT antibody (L20, Santa Cruz Biotechnology Inc, USA) by Western blot analysis and immunofluorescence in combination with telomere FISH.36 Most HeLa and mesothelioma cells, in an asynchronous population containing high levels of telomerase activity, showed bright nuclear speckles (Fig. 6). Similar staining was reported in both asynchronous and synchronized HeLa cells and mouse primary cells, using a T-motif-specific antibody called K370.37 Such a pattern is interesting since it could correspond to replication complexes or to the telomeres. However, whereas no K370 signals were found in telomerase-negative human IMR90 cells, we always observed speckles in telomerase-negative fibroblasts and mesothelial cells with the L20 antibody. In addition, cytoplasmic staining, absence of a specific hTERT band by Western blotting, and lack of foci of colocalization with telomeres in telomerase-positive cells forced us to conclude that the L20 antibody is not suitable for the detection of hTERT protein. These negative results have been confirmed by several other groups (Petra Boukamp, personal communication). Our RT-PCR results discussed above exclude the possibility of cross-reactivity of L20 with translated products of splicing variants.

At the time of writing, other groups reported the successful immunohistochemical detection of hTERT using homemade antibodies. The group of Ide developed the anti-hTERT 1.0 antibody and studied its expression in human colorectal and gastric tumor and nontumor tissue sections. Tahara et al reported the detection of hTERT protein in colonic adenocarcinoma and neoplastic crypt epithelium, where it was confined to the lower portion of the crypts, while Yasui and coworkers detected hTERT protein in the majority of gastric carcinomas but found weak or no staining in non-neoplastic mucosal cells.38,39 The group of Shroyer developed the mouse monoclonal IgG2A kappa antibody 4B1 and studied its expression in cervical and vulvar benign, dysplastic and neoplastic lesions. In cervical lesions, the detection of hTERT seemed to correlate with telomerase activity in high-grade dysplastic lesions (HSIL) and squamous cell carcinomas (SCC), but hTERT was also detected in low grade lesions (LSIL) and normal or benign-reactive cervical mucosa.40 In vulvar intraepithelial neoplasias (VINs), hTERT nuclear staining correlated with squamous maturation and the degree of nuclear atypia.41 More recently, Jerry Shay and coworkers studied the expression of an affinity-purified polyclonal rabbit antibody (EST21A, Alpha Diagnostic International, San Antonio, TX) in several human cancer tissues and a subset of cells in normal tissues.42 The positive hTERT signals were detected as dotted/speckled patterns distributed throughout the nucleus, but were consistently absent from nucleoli. Most importantly, they found that the immunolocalization of hTERT in specimens of adult cancers revealed that the levels of telomerase activity mainly depended on the number of tumor cells with telomerase activity. We are currently testing the EST21A antibody on serosal lesions.
hTERC Antisense Inhibition of Telomerase Function in Mesothelioma Cell Lines
Antisense technology for targeted inhibition of gene expression is growing explosively, and is hoped to offer possibilities for the design of telomerase inhibitors, too. The development of antisense oligonucleotides (ASOs) as therapeutic agents relies on the ability of ASOs to specifically bind to a disease-causing RNA by Watson-Crick base pairing and to directly or indirectly (through recruitment of intracellular RNase H, P, or L) inactivate it. Generally, two routes for the delivery of nucleic acids are employed: (i) transfection with the gene encoding the antisense RNA and (ii) in vitro synthesis of the antisense nucleic acid followed by introduction into the cell. Cellular uptake of exogenously applied nucleic acids is problematic, and different delivery vehicles have been developed. HeLa cells transfected with a hTERC antisense expression construct lost telomeric DNA and began to die after 23 to 26 doublings.43 Incubation of HeLa cells with FuGENE6-encapsulated phosphorothioate ASOs (phosphodiester ASOs with a single oxygen replaced by sulfur) or peptide nucleic acid (PNA)ASOs (modified oligonucleotides with a nonionic backbone) inhibited telomerase in a sequence-selective fashion.44,46
To our knowledge, the effects of anti-telomerase or anti-telomere drugs have never been studied in mesothelioma cancers or mesothelioma cell lines. In collaboration with John K. Cowell (Depts. of Neurosurgery and Neurosciences, The Cleveland Clinic Foundation, Cleveland, Ohio, USA), preliminary in vitro experiments using special phosphorothioate ASOs, called 2-5A antisense oligonucleotides (25-AASOs) were performed47, as described in their paper.48 The ASO, designed against a nontemplate part of hTERC (nucleotides 7694), is covalently attached to four 2',5'-linked tetraadenylate groups (2-5As), which activate endogenous RNase L when the ASO binds to the complementary sequence. As a result, in telomerase-positive cells, hTERC is expected to be degraded after administration of the appropriate 2-5ASOs. In vitro cell viability is shown in Figure 7, as measured by the trypan blue dye exclusion assay, plotted against duration of administration, for 2 of 3 telomerase-positive, mesothelioma-derived cell lines, UIAMM2 and UIAMM7, that were established and characterized in our laboratory.49 In contrast to UIAMM2, UIAMM7 cell viability is reduced to nearly 0% after 6 days of treatment. The results seen with UIAMM7 are encouraging, but the mechanisms behind the effects are unknown. In their series of human malignant glioma cells and tumors, and using the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) technique, the group of Cowell detected a high apoptotic rate in cell cultures and in tumors grown in nude mice.48 Since glioma cells were unlikely to have undergone enough cell divisions to significantly shorten their telomeres, they concluded that telomerase is linked to telomere-independent pathways controlling cell growth rather than to telomere erosion itself. Because we do not have TUNEL data nor data on telomere dynamics during treatment, any conclusion whether the cell death seen with UIAMM7 relies on telomere-dependent or on telomere-independent apoptotic mechanisms is highly speculative. Further, although several controls were included, the question remains to what extent the biological observations obtained with the 2-5ASOs are a function of sequence specificity. The sulphur group in phosphorothioate ASOs renders them much more resistant to nucleolytic degradation than phosphodiester ASOs, but they disadvantageously tend to bind to a large number of proteins, including growth factors, such as basic fibroblast growth factor, platelet-derived growth factor, vascular endothelial growth factor and its receptor, and epidermal growth factor receptor.50 Therefore, it can not be excluded that the observations result from nonsequence-specific mechanisms. In a recent report Matthes and Lehmann similarly found that phosphorothioate ASOs, noncomplementary to hTERC, produced nearly the same inhibitory activity as a complementary one, suggesting a sequence-independent mode of action.51 They demonstrated that the ASO, which was directed against 9 of the 11 bases of the hTERC template region, targeted the TTAGGG binding site of hTERT protein rather than hTERC RNA itself. At present, no information is available to explain the nonresponding nature of UIAMM2 cells. In another report, Cowell and collaborators interestingly found that inhibition of telomerase can alternatively trigger differentiation.52 Whether the 2-5ASO treatment caused UIAMM2 cells to differentiate should be evaluated by monitoring the expression levels of cyclin-dependent kinase inhibitors (p21 and p27) and by motility assays. Also, similar studies need to be repeated, thereby simultaneously measuring transcript expression, telomerase activity and telomere lengths. It is of utmost importance to know whether ALT mechanisms take over telomere stabilization after telomerase inhibition.

Conclusions
Judging by the tally of reported studies and cancer models reviewed by us, telomerase can be regarded as the molecule of the 1990s. Inevitably, as more experimental results are becoming available even more new questions on the biological role and clinical value of telomerase, in carcinogenesis in general and in mesothelioma carcinogenesis in particular, emerge. Nowadays, ‘telomerologists’ can generally be subdivided into 4 schools. Those who regard telomerase as (i) an oncogene,1 (ii) a proliferation marker,11 (iii) a differentiation marker,53 and (iv) a stem cell marker.54 We do not attempt to provide an ultimate answer. However, this study did generate some interesting arguments to join in the aforementioned debate.
The immortalizing telomerase enzyme is considered to be an oncoprotein based on the hypothesis that the mortal phenotype, that characterizes most somatic cells represents a potent tumor suppressor mechanism, and on the finding that nearly all malignant cells/tumors have telomerase activity. We were able to describe the telomerase status at the ends of the spectrum of mesothelioma carcinogenesis, and indeed found that telomerase closely associates with the malignant phenotype.
Furthermore, in our opinion, the lack of telomerase activity and hTERT expression observed in mesothelial cell cultures can be extrapolated to the in vivo setting. Indeed, the in vivo telomerase-incompetent nature of certain cell types is only acceptable when such cell types do not express telomerase in vitro.15 It should be mentioned that the absence of a trait is always difficult to prove. Therefore, we can not exclude that normal mesothelial cells do express telomerase under other in vitro culturing conditions. To the present, and because nearly all malignant mesotheliomas have telomerase activity and express hTERT, it is therefore concluded that telomerase is reactivated, rather than up-regulated, during mesothelioma carcinogenesis.
A central question concerns the nature of the mechanisms or of the agents, which cause telomerase reactivation. According to Belair et al, telomerase activity is regulated by proliferation processes.14 However, in previous work on melanoma carcinogenesis, we excluded that telomerase activity is an epiphenomenon induced by the proliferation machinery, at least in proliferating melanocytes.16 A similar conclusion was recently drawn for human B lymphocytes.55 On the other hand, it can not be denied that a close association between telomerase activity and proliferation does exist in several other human cell types such as endometrial cells, trophoblasts and haemotopoietic progenitors.56,57 It is currently rather mysterious why both parameters coincide in some tissues and not in others. An answer may come from studies investigating the hTERT promotor. Indeed, the c-myc (proto)oncoprotein, for which E-box binding sites are present on the hTERT promotor, can activate hTERT transcription but also induces cell cycle progression via indirect activation of cyclin E/Cdk2.58 However, Kyo et al recently found that c-myc is only transcriptionally active when Sp1 is expressed at a critical level, and concurrently binds to one or more GC-boxes within the hTERT promotor.59 Substantial variations in expression of this transacting transcription factor Sp1 have been found in several cell types.60 Therefore, it seems important to determine Sp1 expression by e.g., Western blotting in cultured mesothelial cells compared to trophoblasts, melanomas or mesotheliomas.
In search for a positive answer to the aforementioned central question, one should focus on those processes, which lead to lifespan extension and concomitant telomere erosion. It was found that, after transfection with a plasmid encoding the SV40 early region, normal human mesothelial cells (Met4A and Met5A cell lines) acquire an extended lifespan, and eventually become immortalized and have telomerase activity.61 We and others found SV40 DNA-like sequences in our mesothelioma series.20 The study of telomerase and SV40 in mesothelioma is interesting for at least 3 other reasons. Weinberg and collaborators recently reported the first successful creation of human tumor cells with defined genetic elements (hTERT, SV40 LTag and Hras), demonstrating that disruption of the intracellular pathways regulated by the telomerase catalytic protein, the large-T and the H-ras oncogene suffices to create a human tumor cell.62 So, telomere maintenance, facilitated by hTERT expression, in vivo might cooperate with additional oncogenic mutations to create a malignantly transformed clone. Therefore, it is fascinating to know if, besides SV40 LTag and hTERT, a third player also suffices to trigger the malignant transformation of the mesothelium, and how this player looks like. Indeed, Ramael et al and Segers et al already demonstrated that H-ras mutations and protein expression in malignant mesothelioma is not frequent.63,64 Since immortal SV40-transfected cell lines as well as cell lines expressing the N-ras and H-ras oncogenes assign to the same Complementation Group A, as defined by Pereira-Smith and Smith in 198865, this third player could be looked for among those factors typical for the Complementation Group A. Secondly, the SV40 LTag was found to be able to unwind fourstranded DNA structures linked by stacked G-quartets.66 It needs further study to find out whether this ‘helicase’ activity may affect processes such as telomere extension, in which four-stranded DNA could play a role. Tertio, cancer cells are generally recognized as being in a relatively undifferentiated state and could hypothetically arise either from damaged somatic cells or from stem cells with blocked or partially blocked differentiation.67 A supposed origin of SFTs or mesotheliomas from subserosal stem cells, as stated by some authors, follows the latter hypothesis. Interestingly, Sun et al recently found that human breast epithelial cells with stem cell characteristics (HBECs Type I) are more susceptible to telomerase activation and immortalisation after transfection with SV40 LTag than commercially SV40-transfected HBECs without stem cell characteristics (HEBCs Type II).68 Transferring this observation to mesothelioma, it could be that subserosal stem cells deserve much more attention in mesotheliomogenesis, in that they may well be the most important targets for the SV40 virus, and the real mesothelioma progenitor cells.
For the SV40-negative mesotheliomas, the abovementioned discussion does not seem to hold at first glance. However, as documented for the HPV-16 E6 and E7 immortalized mesothelial cell line MePV23I, it is possible for virally immortalized cell clones to continue proliferating indefinitely, even after deletion of the viral genes.61 In such cases, loss of p16ink4 by homozygous deletion of the gene was found to have substituted for viral oncoprotein-mediated inactivation of pRB. Although such an event is a rarity, it would be interesting to know whether SV40-negative mesotheliomas lost p16 expression, either by homozygous deletion of the gene, small deletions, point mutations or de novo methylation of its 5' CpG islands. Alternatively, future research may discover the presence of (yet unknown) viruses in mesothelioma samples. A recent report did not detect DNA sequences of the human herpesvirus 8 (HHV8) in mesothelioma.69
Recently, the group of Gerwin demonstrated that a subset (25%) of normal human mesothelial cells from individual donors can develop an extended proliferative lifespan in response to direct asbestos (amosite) treatment in vitro, although, for most cells, asbestos treatment leads to cell death through apoptosis or cytotoxicity.70 Unfortunately, the authors, who concentrated in the first place on the expression of the p16ink4 gene during this lifespan extension, did look neither for asbestos-induced generation of immortalized clones nor for asbestos-induced telomerase activity.
As to its biological significance, it is our opinion that it is currently unclear whether telomerase reactivation is a programmed or an accidental event. According to the former hypothesis, gradual telomere erosion forces a cell to activate a telomere-stabilizing rescue mechanism. Indeed, the exhaustion of telomeric buffer, by exposure to asbestos fibers or to oncoviruses, leads to telomeric instability, that is characterized by the presence of a variety of telomeric associations, and it is believed that they cause deletions and structural rearrangements as the result of breakage fusion-bridge cycles between chromosomes, resulting in lethal losses of chromosomal material.71 Unfortunately, while preventing cell death, the reactivation of telomerase opens the door for the further propagation of damaged DNA, and the generation of malignant clones. According to the latter hypothesis, telomerase is pathologically reactivated indirectly by telomeric associations-induced loss of telomerase repressors, or directly by the impact of asbestos fibers or viral oncoproteins upon the hTERT promotor. Further research definitely will provide an answer to these intriguing questions.
Future Perspectives
As clinically oriented morphologists, we are aware that this work did not yet entirely succeed in answering the final question whether telomerase activity is able to differentiate benign from malignant mesothelial proliferations. This is mainly due to the scarcity of fresh or frozen serosal materials. In this respect, we mention the publishing of a recent report that showed no telomerase activity in 5 reactive pleural cytology specimens. Unfortunately, a histologic diagnosis was not available.72
As stated in our recent review,73 ectopic expression of hTERT may offer possibilities to test models of mesothelioma carcinogenesis using normal human mesothelial cells, which otherwise are rather difficult to culture. We therefore embrace a very recent article, published by the group of Rheinwald, reporting that (peritoneal) mesothelial cells (strain LP9) could be immortalized by solely introduction of hTERT, and, more importantly, this strain LP9TERT1 was not accompanied by alterations of growth or differentiation characteristics.74 One may hope that this cell line LP9TERT1 soon becomes commercially available.
References
- 1.
- Kim NW, Piatyszek MA, Prowse KR. et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. [PubMed: 7605428]
- 2.
- Wright WE, Shay JW, Piatyszek MA. Modifications of a telomeric repeat amplification protocol (TRAP) result in increased reliability, linearity and sensitivity. Nucleic Acids Res. 1995;23:3794–3795. [PMC free article: PMC307284] [PubMed: 7479015]
- 3.
- Krupp G, Kühne K, Tamm S. et al. Molecular basis of artifacts in the detection of telomerase activity and a modified primer for a more robust ‘TRAP’ assay. Nucleic Acids Res. 1997;25:919–921. [PMC free article: PMC146494] [PubMed: 9016650]
- 4.
- Kim NW, Wu F. Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP). Nucleic Acids Res. 1997;25:2595–2597. [PMC free article: PMC146790] [PubMed: 9185569]
- 5.
- Tatematsu K, Nakayama J, Danbara M. et al. A novel quantitative ‘stretch PCR assay’, that detects a dramatic increase in telomerase activity during the progression of myeloid leukemias. Oncogene. 1996;13:2265–2274. [PubMed: 8950994]
- 6.
- Dhaene K, Hübner R, Kumar-Singh S. et al. Telomerase activity in human pleural mesothelioma. Thorax. 1998;53:915–918. [PMC free article: PMC1745102] [PubMed: 10193387]
- 7.
- Lechner JF, Tesfaigzi J, Gerwin BI. Oncogenes and tumor-suppressor genes in mesothelioma--a synopsis. Environ Health Perspect. 1997;105 Suppl 5:1061–1067. [PMC free article: PMC1470150] [PubMed: 9400701]
- 8.
- Craighead JE. Current pathogenetic concepts of diffuse malignant mesothelioma. Hum Pathol. 1987;18:544–557. [PubMed: 3036684]
- 9.
- Shay JW, Wright WE. The reactivation of telomerase activity in cancer progression. Trends Genet. 1996;12:129–131. [PubMed: 8901415]
- 10.
- Greaves M. Is telomerase activity in cancer due to selection of stem cells and differentiation arrest. Trends Genet. 1996;12:127–128. [PubMed: 8901414]
- 11.
- Greider CW. Telomerase activity, cell proliferation, and cancer. Proc Natl Acad Sci USA. 1998;95:90–92. [PMC free article: PMC34198] [PubMed: 9419332]
- 12.
- Sallinen P, Miettinen H, Sallinen SL. et al. Increased expression of telomerase RNA component is associated with increased cell proliferation in human astrocytomas. Am J Pathol. 1997;150:1159–1164. [PMC free article: PMC1858183] [PubMed: 9094971]
- 13.
- Ogoshi M, Le T, Shay JW. et al. In situ hybridization analysis of the expression of human telomerase RNA in normal and pathologic conditions of the skin. J Invest Dermatol. 1998;110:818–823. [PubMed: 9579552]
- 14.
- Belair CD, Yeager TR, Lopez PM. et al. Telomerase activity: a biomarker of cell proliferation, not malignant transformation. Proc Natl Acad Sci USA. 1997;94:13677–13682. [PMC free article: PMC28365] [PubMed: 9391085]
- 15.
- Wynford-Thomas D. Cellular senescence and cancer. J Pathol. 1999;187:100–111. [PubMed: 10341711]
- 16.
- Dhaene K, Vancoillie G, Lambert J. et al. Absence of telomerase activity and telomerase catalytic subunit mRNA in melanocyte cultures. Br J Cancer. 2000;82:1051–1057. [PMC free article: PMC2374429] [PubMed: 10737388]
- 17.
- Rudolph P, Knüchel R, Endl E. et al. The immunohistochemical marker Ki-S2: cell cycle kinetics and tissue distribution of a novel proliferation-specific antigen. Mod Pathol. 1998;11:450–456. [PubMed: 9619598]
- 18.
- Umbricht CB, Saji M, Westra WH. et al. Telomerase activity: a marker to distinguish follicular thyroid adenoma from carcinoma. Cancer Res. 1997;57:2144–2147. [PubMed: 9187112]
- 19.
- Counter CM, Avilion AA, LeFeuvre CE. et al. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. [PMC free article: PMC556651] [PubMed: 1582420]
- 20.
- Dhaene K, Verhulst A, Van Marck E. SV40 large T-antigen and human pleural mesothelioma. Screening by polymerase chain reaction and tyramine-amplified immunohistochemistry. Virchows Arch. 1999;435:1–7. [PubMed: 10431839]
- 21.
- Carbone M, Rizzo P, Pass HI. Simian virus 40, poliovaccines and human tumors: a review of recent developments. Oncogene. 1997;15:1877–1888. [PubMed: 9365233]
- 22.
- Kipling D. Telomere structure and telomerase expression during mouse development and tumorigenesis. Eur J Cancer. 1997;33:792–800. [PubMed: 9282119]
- 23.
- Wynford-Thomas D, Kipling D. Telomerase. Cancer and the knockout mouse. Nature. 1997;389:551–552. [PubMed: 9335490]
- 24.
- Nakamura TM, Morin GB, Chapman KB. et al. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. [PubMed: 9252327]
- 25.
- Nakayama J, Tahara H, Tahara E. et al. Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat Genet. 1998;18:65–68. [PubMed: 9425903]
- 26.
- Ramakrishnan S, Eppenberger U, Mueller H. et al. Expression profile of the putative catalytic subunit of the telomerase gene. Cancer Res. 1998;58:622–625. [PubMed: 9485011]
- 27.
- Ulaner GA, Hu JF, Vu TH. et al. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 1998;58:4168–4172. [PubMed: 9751630]
- 28.
- Dhaene K, Wauters J, Weyn B. et al. Expression profile of telomerase subunits in human pleural mesothelioma. J Pathol. 2000;190:80–85. [PubMed: 10640996]
- 29.
- Cong YS, Wen JP, Bacchetti S. The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum Mol Genet. 1999;8:137–142. [PubMed: 9887342]
- 30.
- Takakura M, Kyo S, Kanaya T. et al. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999;59:551–557. [PubMed: 9973199]
- 31.
- Greenberg RA, O'Hagan RC, Deng H. et al. Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene. 1999;18:1219–1226. [PubMed: 10022128]
- 32.
- Wu KJ, Grandori C, Amacker M. et al. Direct activation of TERT transcription by c-Myc. Nat Genet. 1999;21:220–224. [PubMed: 9988278]
- 33.
- Ramael M, Van Den Bossche J, Buysse C. et al. Immunoreactivity for c-fos and c-myc protein with the monoclonal antibodies 14E10 and 6E10 in malignant mesothelioma and non-neoplastic mesothelium of the pleura. Histol Histopathol. 1995;10:639–643. [PubMed: 7579812]
- 34.
- Cuthbert AP, Bond J, Trott DA. et al. Telomerase repressor sequences on chromosome 3 and induction of permanent growth arrest in human breast cancer cells. J Natl Cancer Inst. 1999;91:37–45. [PubMed: 9890168]
- 35.
- Lee WC, Testa JR. Somatic genetic alterations in human malignant mesothelioma (review). Int J Oncol. 1999;14:181–188. [PubMed: 9863027]
- 36.
- Dhaene K, Wauters J, Weyn B. et al. Expression profile of telomerase subunits in human pleural mesothelioma. J Pathol. 2000;190:80–85. [PubMed: 10640996]
- 37.
- Martin-Rivera L, Herrera E, Albar JP. et al. Expression of mouse telomerase catalytic subunit in embryos and adult tissues. Proc Natl Acad Sci USA. 1998;95:10471–10476. [PMC free article: PMC27918] [PubMed: 9724727]
- 38.
- Tahara H, Yasui W, Tahara E. et al. Immuno-histochemical detection of human telomerase catalytic component, hTERT, in human colorectal tumor and non-tumor tissue sections. Oncogene. 1999;18:1561–1567. [PubMed: 10102626]
- 39.
- Yasui W, Tahara H, Tahara E. et al. Expression of telomerase catalytic component, telomerase reverse transcriptase, in human gastric carcinomas. Jpn J Cancer Res. 1998;89:1099–1103. [PMC free article: PMC5921711] [PubMed: 9914776]
- 40.
- Frost M, Bobak JB, Gianani R. et al. Localisation of telomerase hTERT protein and hTR in benign mucosa, dysplasia, and squamous cell carcinoma of the cervix. Am J Clin Pathol. 2000;114:726–734. [PubMed: 11068546]
- 41.
- Wada H, Enomoto T, Yoshino K. et al. Immunohistochemical localisation of telomerase hTERT protein and analysis of clonality in multifocal vulvar intraepithelial neoplasia. Am J Clin Pathol. 2000;114:371–379. [PubMed: 10989637]
- 42.
- Hiyama E, Hiyama K, Yokoyama T. et al. Immunohistochemical detection of telomerase (hTERT) protein in human cancer tissues and a subset of cells in normal tissues. Neoplasia. 2001;3:17–26. [PMC free article: PMC1505023] [PubMed: 11326312]
- 43.
- Feng J, Funk WD, Wang SS. et al. The RNA component of human telomerase. Science. 1995;269:1236–1241. [PubMed: 7544491]
- 44.
- Tao M, Miyano-Kurosaki N, Takai K. et al. Specific inhibition of human telomerase activity by transfection reagent, FuGENE6-antisense phosphorothioate oligonucleotide complex in HeLa cells. FEBBS Lett. 1999;454:312–316. [PubMed: 10431829]
- 45.
- Hamilton SE, Pitts AE, Katipally RR. et al. Identification of determinants for inhibitor binding within the RNA active site of human telomerase using PNA scanning. Biochemistry. 1997;36:11873–11880. [PubMed: 9305980]
- 46.
- Norton JC, Piatyszek MA, Wright WE. et al. Inhibition of human telomerase activity by peptide nucleic acids. Nat Biotechnol. 1996;14:615–619. [PubMed: 9630953]
- 47.
- Dhaene K, Van Marck E. Telomerase function in the diagnosis and treatment of malignant mesotheliomaIn: Peters GA, BJ Peters, eds.Sourcebook on Asbestos Diseases LEXIS Law Publishing,2000 .
- 48.
- Kondo S, Kondo Y, Li G. et al. Targeted therapy of human malignant glioma in a mouse model by 2-5A antisense directed against telomerase RNA. Oncogene. 1998;16:3323–3330. [PubMed: 9681832]
- 49.
- KumarSingh S. Differentiation and progression of malignant mesothelioma 1998. University of Antwerp. Thesis . [PMC free article: PMC140111]
- 50.
- Stein CA. Phosphorothioate antisense oligodeoxynucleotides:questions of specificity. Trends Biotechnol. 1996;14:147–149. [PubMed: 8645447]
- 51.
- Matthes E, Lehmann Ch. Telomerase protein rather than its RNA is the target of phosphorothioate-modified oligonucleotides. Nucleic Acids Res. 1999;27:1152–1158. [PMC free article: PMC148297] [PubMed: 9927750]
- 52.
- Kondo S, Tanaka Y, Kondo Y. et al. Antisense telomerase treatment: induction of two distinct pathways, apoptosis and differentiation. FASEB J. 1998;12:801–811. [PubMed: 9657520]
- 53.
- Kipling D. Mammalian telomerase: catalytic subunit and knockout mice. Hum Mol Genet. 1997;6:1999–2004. [PubMed: 9328462]
- 54.
- Härle-Bachor C, Boukamp P. Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes. Proc Natl Acad Sci USA. 1996;93:6476–6481. [PMC free article: PMC39048] [PubMed: 8692840]
- 55.
- Hu BT, Insel RA. Up-regulation of telomerase in human B lymphocytes occurs independently of cellular proliferation and with expression of the telomerase catalytic subunit. Eur J Immunol. 1999;29:3745–3753. [PubMed: 10556831]
- 56.
- Bonatz G, Klapper W, Barthe A, Heidorn K, Jonat W, Krupp G, Parwaresch R. Analysis of telomerase expression and proliferative activity in the different layers of cyclic endometrium. Biochem Biophys Res Commun. 1998;253:214–221. [PubMed: 9878518]
- 57.
- Kyo S, Takakura M, Tanaka M. et al. Expression of telomerase activity in human chorion. Biochem Biophys Res Commun. 1997;241:498–503. [PubMed: 9425299]
- 58.
- Bouchard C, Staller P, Eilers M. Control of cell proliferation by Myc. Trends Cell Biol. 1998;8:202–206. [PubMed: 9695840]
- 59.
- Kyo S, Takakura M, Taira T. et al. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res. 2000;28:669–677. [PMC free article: PMC102554] [PubMed: 10637317]
- 60.
- Saffer JD, Jackson SP, Annarella MB. Developmental expression of Sp1 in the mouse. Mol Cell Biol. 1991;11:2189–2199. [PMC free article: PMC359911] [PubMed: 2005904]
- 61.
- Noble JR, Rogan EM, Neumann AA. et al. Association of extended in vitro proliferative potential with loss of p16INK4 expression. Oncogene. 1996;13:1259–1268. [PubMed: 8808700]
- 62.
- Hahn WC, Counter CM, Lundberg AS. et al. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. [PubMed: 10440377]
- 63.
- Ramael M, Deblier I, Eerdekens C. et al. Immunohistochemical staining of ras oncogene product in neoplastic and non-neoplastic mesothelial tissues: immunoreactivity for N-ras and lack of immunohistochemical staining for Ha-ras and K-ras. J Pathol. 1993;169:421–424. [PubMed: 8501538]
- 64.
- Segers K. Study of DNA changes and of growth related proteins in malignant mesothelioma 1996. University of Antwerp. Thesis. [PMC free article: PMC172907]
- 65.
- Pereira-Smith OM, Smith JR. Genetic analysis of indefinite division in human cells: identification of four complementation groups. Proc Natl Acad Sci USA. 1988;85:6042–6046. [PMC free article: PMC281901] [PubMed: 3413074]
- 66.
- Baran N, Pucshansky L, Marco Y. et al. The SV40 large T-antigen helicase can unwind four stranded DNA structures linked by Gquartets. Nucleic Acids Res. 1997;25:297–303. [PMC free article: PMC146434] [PubMed: 9016557]
- 67.
- Varmus HE, Weinberg RA. The nature of cancerIn: Varmus HE, RA Weinberg, eds.Genes and the biology of cancerScientific American Library,199338–39.
- 68.
- Sun W, Kang KS, Morita I. et al. High susceptibility of a human breast epithelial cell type with stem cell characteristics to telomerase activation and immortalization. Cancer Res. 1999;59:6118–6123. [PubMed: 10626801]
- 69.
- Ascoli V, Nardi F, Carnovale Scalzo C. et al. Absence of HHV-8 DNA sequences in malignant mesothelioma. Mol Pathol. 1998;51:113–114. [PMC free article: PMC395621] [PubMed: 9713597]
- 70.
- Xu L, Flynn BJ, Ungar S. et al. Asbestos induction of extended lifespan in normal human mesothelial cells: interindividual susceptibility and SV40 T antigen. Carcinogenesis. 1999;20:773–783. [PubMed: 10334193]
- 71.
- Hastie ND, Allshire RC. Human telomeres: fusion and interstitial sites. Trends Genet. 1989;5:326–331. [PubMed: 2692239]
- 72.
- Mu XC, Brien TP, Ross JS. et al. Telomerase activity in benign and malignant cytologic fluids. Cancer. 1999;87:93–99. [PubMed: 10227600]
- 73.
- Dhaene K, Van Marck E, Parwaresch R. Telomeres, telomerase and cancer: an update. Virchows Arch. 2000;437:1–16. [PubMed: 10963374]
- 74.
- Dickson MA, Hahn WC, Ino Y. et al. Human keratinocytes that express hTERT and also bypass a p16INK4a-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 2000;20:1436–1447. [PMC free article: PMC85304] [PubMed: 10648628]
- Abbreviations
- Introduction
- Telomerase Activity in Malignant Mesothelioma
- Gene Expression Profile of Components of Telomerase in Mesothelioma
- Detection of Telomerase at the Cellular Level
- hTERC Antisense Inhibition of Telomerase Function in Mesothelioma Cell Lines
- Conclusions
- Future Perspectives
- References
- Telomerase in Mesothelioma: Diagnostic and Therapeutic Applications - Madame Cur...Telomerase in Mesothelioma: Diagnostic and Therapeutic Applications - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...