Methods for the synthesis of evidence of clinical effectiveness
A systematic review of the clinical effectiveness of adalimumab, etanercept and ustekinumab within their respective licensed indications for the treatment of plaque psoriasis in children and young people was performed following the general principles recommended in the Centre for Reviews and Dissemination (CRD)’s guidance28 and the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement.29 A protocol was registered with PROSPERO.
Literature searching: adalimumab, etanercept and ustekinumab
The literature search for the clinical effectiveness review aimed to systematically identify relevant RCTs of adalimumab, etanercept and ustekinumab used to treat children and young people with plaque psoriasis.
The search strategy was developed in MEDLINE (via Ovid) and included search terms for:
psoriasis
adalimumab, etanercept, ustekinumab or biosimilars
children or young people.
The three sets of terms were combined using the Boolean operator AND. Search terms were developed through discussion with the review team and use of database thesauri and online drug information resources. No language, date, geographical or study design limits were applied. The MEDLINE strategy was adapted for use in the other resources searched.
The searches were carried out on 24/25 May 2016 and updated during September 2016. The following databases were searched: MEDLINE (including MEDLINE Epub Ahead of Print, MEDLINE In-Process & Other Non-Indexed Citations, Ovid MEDLINE Daily and Ovid MEDLINE), Cochrane Central Register of Controlled Trials (CENTRAL), Cochrane Database of Systematic Reviews (CDSR), Cumulative Index to Nursing and Allied Health Literature (CINAHL) Plus, Database of Abstracts of Reviews of Effects (DARE), EMBASE, Health Technology Assessment (HTA) database, NHS Economic Evaluation Database (NHS EED), PubMed and Science Citation Index.
In addition, the following resources were searched for ongoing, unpublished or grey literature: ClinicalTrials.gov, Conference Proceedings Citation Index – Science, EU Clinical Trials Register, PROSPERO and World Health Organization (WHO) International Clinical Trials Registry Platform portal.
A search for guidelines on psoriasis in children or young people was carried out through the following guideline websites: National Guideline Clearinghouse (www.guideline.gov; accessed 14 June 2017), NICE Clinical Knowledge Summaries (https://cks.nice.org.uk/; accessed 14 June 2017), NHS Evidence (www.evidence.nhs.uk; accessed 14 June 2017), NICE evidence summaries: new medicines (www.evidence.nhs.uk/Search?q=Evidence+summary+new+medicine; accessed 14 June 2017) and the NICE website (www.nice.org.uk/; accessed 14 June 2017).
In addition to utilising these published and unpublished data resources, requests for clinical study reports (CSRs) relating to adalimumab, etanercept and ustekinumab were made to AbbVie, Pfizer and Janssen respectively.
The search results were imported into EndNote X7 (Thomson Reuters, CA, USA) and deduplicated. Full search strategies can be found in Appendix 1.
Literature searching: network meta-analysis
Alternative treatments in children and young people
To inform the network meta-analysis (NMA), searches were undertaken to identify relevant RCTs of systemic non-biological (acitretin, methotrexate and ciclosporin) and other biological (infliximab, secukinumab) therapies used in children and young people with plaque psoriasis. No language, date, geographical or study design limits were applied to the searches.
The searches were carried out on 31 May 2016 in the following databases: MEDLINE (including MEDLINE Epub Ahead of Print, MEDLIINE In-Process & Other Non-Indexed Citations, Ovid MEDLINE Daily and Ovid MEDLINE), CENTRAL, CDSR, CINAHL Plus, DARE, EMBASE, HTA database, PubMed and Science Citation Index.
In addition, the following resources were searched for ongoing, unpublished or grey literature: ClinicalTrials.gov, Conference Proceedings Citation Index – Science, EU Clinical Trials Register, PROSPERO and WHO International Clinical Trials Registry Platform portal.
The search results were imported into EndNote X7 and deduplicated. The search was updated in September 2016 to capture more recent studies. Full search strategies can be found in Appendix 1.
Registry data
To identify longer-term follow-up evidence, a literature search was conducted within the MEDLINE database for the search terms ‘psoriasis AND regist*’. The results of this search were screened for publications from psoriasis registries, secondary analyses of registry data and systematic reviews of broader dermatological and psoriasis registry data. The list of registries generated through these searches was compared against those in three relevant systematic reviews30–32 to verify the studies included and to identify any that had been overlooked. Twenty patient registries for psoriasis treatment were identified in this way; 14 were located in European countries, three were international in scope, two were based in the USA and one was based in Malaysia. Each registry name was then separately used as a search term in MEDLINE and any publications referencing these that had not been found in the initial searches were retrieved.
In addition, representatives of the 14 psoriasis registries from European countries (Austria, Australia, Czech Republic, Denmark, France, Germany, Italy, Netherlands, Portugal, Slovenia, Spain, Sweden, Switzerland and the UK) were contacted and asked to provide any relevant information on the use of the biologics adalimumab, etanercept and ustekinumab for the treatment of psoriasis in children and young people.
Inclusion and exclusion criteria
Two reviewers independently screened all titles and abstracts. Full manuscripts for any potentially relevant titles/abstracts were obtained when possible and the relevance of each study was assessed by two reviewers according to the following criteria. Any discrepancies were resolved by consensus and, if necessary, a third reviewer was consulted. Studies available only as abstracts were included and attempts were made to contact the authors for further details.
Study design
Randomised controlled trials (including any open-label extensions of RCTs) were eligible for the review of clinical efficacy.
Information on adverse events (AEs) was also sought from regulatory sources when appropriate. Registries and observational studies were included when relevant outcome data were available.
To address longer-term measures of efficacy and drug survival, published analyses based on large and long-term data sets (including studies of registry data) were also considered.
Participants
Studies of children and/or young people with moderate to severe plaque psoriasis were included. Severity could be defined using the PASI, PGA, BSA or other measures, alone or in combination, although there is no universal definition of severity for this population. Studies of guttate, erythrodermic and pustular psoriasis were excluded, as were studies of psoriatic arthritis.
Studies in children or young people with psoriasis in whom topical therapies, systemic therapies or phototherapies were inadequate, inappropriate or not tolerated were eligible for inclusion. Participants aged < 12 years were considered to be children whereas those aged 12–17 years were considered to be young people.
Interventions
The relevant interventions were adalimumab, etanercept and ustekinumab.
Comparators
The relevant comparators were:
alternative biological therapies with relevant marketing authorisation (adalimumab, etanercept or ustekinumab)
non-biological systemic therapy (including, but not limited to, ciclosporin and methotrexate)
topical therapy (for people in whom non-biological systemic therapy is not suitable), that is, best supportive care (BSC)
biological treatments used outside their marketing authorisation (such as infliximab, adalimumab, etanercept or ustekinumab if used outside the constraints of the relevant marketing authorisation in children and young people)
biosimilars of etanercept, adalimumab or ustekinumab
placebo.
Outcomes
Data on effectiveness, adverse effects, patient-centred outcome measures, costs to the health service and cost-effectiveness were eligible for inclusion, including the following outcomes:
severity of psoriasis (e.g. BSA, PGA score)
response and remission rates (e.g. PASI 50/75/90 response)
relapse rate
rates of treatment discontinuation and withdrawal
short- and long-term adverse effects of treatment (e.g. injection site and allergic reactions, serious infections, reactivation of infections including tuberculosis, malignancy)
HRQoL [e.g. CDLQI, PedsQL and EuroQol-5 Dimensions (EQ-5D)
33 scores].
Data extraction
Data relating to both study design and study quality were extracted by one reviewer using a standardised data extraction form and independently checked for accuracy by a second reviewer. Disagreements were resolved through consensus and, if necessary, a third reviewer was consulted. Data from studies with multiple publications were extracted and reported as a single study.
Quality assessment
The quality of RCTs was assessed using the Cochrane risk-of-bias tool,34 with additional assessments made for baseline imbalance of important prognostic indicators.35 Relevant prognostic and treatment response indicators were identified from both published research and clinical advice. The risk-of-bias assessment was performed by one reviewer and independently checked by a second. Disagreements were resolved through consensus and, if necessary, a third reviewer was consulted.
The quality of non-randomised studies was assessed using a checklist based on CRD guidance28 and used in previous technology assessments for NICE.36 This assesses study eligibility criteria and recruitment methods, the baseline similarity of comparison groups, the blinding of allocation, the completeness of follow-up and outcome reporting.
Methods of data synthesis
The analysis and synthesis of clinical data in this review was conducted in distinct sections. In the absence of sufficient trials to conduct pairwise meta-analysis, the results of included studies are presented in a series of structured tables and summarised narratively and subjected to detailed critical appraisal.
To assess the relative clinical effectiveness of the three biologics (i.e. adalimumab, etanercept and ustekinumab), syntheses of both pairwise (head-to-head) and indirect comparative data were planned. When possible, treatment response (PASI) outcomes were to be synthesised using Bayesian NMA methods. Bayesian statistical methods provide information on the benefits of the active treatments relative to the appropriate comparators and each other.37 Meta-analysis using mixed-treatment comparisons enables the estimation of different parameters from several studies with similar comparisons to be combined when direct evidence on comparisons of interest is absent or sparse.38 For example, should active treatments being evaluated have a common comparator of placebo, this would allow a network to be established between them, providing information on the benefits of these treatments relative to placebo and to each other.
However, the available trials conducted in children precluded the construction of the necessary network. To inform the economic evaluation, trials conducted in adults were included in a NMA. Full details of the methods and results are presented in Chapter 4 (see Framework of analysis for informing the relative efficacy of the interventions).
Results
Quantity of identified evidence
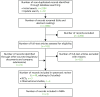
A total of 2386 non-duplicate records were identified from the clinical effectiveness database searches. Of these, 2284 records were excluded after title or abstract screening. In addition, eight relevant regulatory documents were retrieved. Thus, a total of 111 records were read in full, resulting in 63 records being excluded and a total of 48 records being included in the review,39–86 relating to nine studies (three RCTs and six open-label or observational follow-up studies) (). The included records are summarised in Appendix 2. Appendix 3 lists the excluded studies and reasons for exclusion.
Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram of studies in children and young people.
Searches for relevant registry data identified 685 publications. Three publications from two registries were found to include children with psoriasis who were treated with biologics. Of the 14 national psoriasis registry representatives contacted, seven responded but no relevant additional data were available.
Characteristics of the included studies
Three RCTs were retrieved, one for each of the biologics of interest (i.e. adalimumab,39–47,79,80,87 etanercept48–69 and ustekinumab72–75,81,82,88). The RCTs investigated short-term clinical efficacy and AEs. The etanercept and ustekinumab trials included 12 weeks of follow-up and used placebo as a comparator whereas the adalimumab trial was of 16 weeks’ duration and included oral methotrexate, a non-biological systemic treatment, as the comparator. Participant selection criteria for these trials are reported in . Each RCT also incorporated an open-label phase (). These open-label or observational periods investigated longer-term efficacy and AEs, incorporating withdrawal and/or retreatment phases. The adalimumab, etanercept and ustekinumab trials had 52, 312 and 60 weeks of follow-up data available respectively.
Inclusion and exclusion criteria for included RCTs
Trial durations (including open-label extensions) and dosing regimens
Baseline characteristics of participants
The baseline characteristics of the participants in the included RCTs are presented in . Although only older children and adolescents (aged 12–17 years) were included in the ustekinumab trial, the median age of children across the three trials did not differ greatly, as it appears that relatively few younger children were included in the adalimumab and etanercept trials.
Baseline characteristics of participants in the included RCTs
All three trials used a composite measure of disease severity incorporating baseline PASI, PGA and BSA measurements. When used in isolation, a PASI score between 10 and 20 is considered to indicate moderate to severe psoriasis, whereas severe psoriasis has a score of > 20. Across the included studies, the average PASI score ranged from 18.3 to 21.2, with 93–100% of participants having a PGA score of > 3 (mild/moderate disease). Although adalimumab and etanercept are licensed for severe chronic plaque psoriasis and ustekinumab is licensed for moderate to severe plaque psoriasis, on average, measures of disease duration and the component measures of severity did not appear to differ markedly between the three trials. The degree of psoriasis affecting high-impact and difficult-to-treat sites (e.g. face, scalp, palms, soles, flexures and genitals) across the three studies was less clear.
A key difference between the licences for the three agents is the availability of adalimumab for patients for whom topical therapy and phototherapy are inadequate or inappropriate. Unlike the licences for etanercept and ustekinumab, there is no mention in the licence for adalimumab of previous non-biological systemic treatment. However, a substantial minority of participants in the adalimumab trial (29.8%) had received prior systemic therapy, compared with 42.7% of participants in the ustekinumab trial; 56.8% of participants in the etanercept trial had received either prior systemic therapy or prior phototherapy (separate data were not reported).
A similar proportion of participants in the adalimumab and ustekinumab trials had received some form of biological treatment prior to enrolment (9.6% and 10.8% respectively). As etanercept was the first TNF-α inhibitor to be approved for psoriasis, none of the participants recruited to the etanercept trial had previously been treated with biological therapy.
Although there were noticeable differences in participant characteristics between the trials, these were not as clear as the respective licences for the three treatments might suggest. Notwithstanding methodological differences, there appears to be sufficient overlap in the trial populations to discuss these three trials together.
Length of follow-up and early escape
The initial randomised treatment period was 12 weeks in the etanercept and ustekinumab trials and 16 weeks in the adalimumab trial. Twelve-week outcome data were not available for the adalimumab trial, although clinical advice (Dr Ruth Murphy, Consultant Dermatologist, Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield, 25 October 2016, personal communication) suggested that the difference in length of follow-up between treatments was acceptable.
All three trials allowed participants to ‘escape’ from the randomised treatment period before the 12-/16-week follow-up. The criteria for early escape and statistical handling of early escape data are discussed separately for each trial in the relevant sections.
Post-randomised treatment periods are briefly summarised in .
Outcomes
The adalimumab and etanercept trials considered the PASI 75 response to be the primary outcome measure, whereas the ustekinumab trial used a primary outcome measure of a PGA score of 0 or 1 (‘clear’ or ‘almost clear’). However, all three trials reported PASI and PGA scores and some measure of HRQoL (CDLQI and/or PedsQL), which are presented in the following sections.
Efficacy and safety of adalimumab
One multicentre RCT (M04-717) comparing two doses of adalimumab against methotrexate met the selection criteria for the review. Although this trial has not been published in a peer-reviewed journal, data were available from regulatory documentation,46,47 conference proceedings39–44,87 and a CSR provided by the manufacturer.45
The M04-717 trial was separated into four periods:
period A – double-blind RCT of initial treatment (16 weeks)
period B – observational study of treatment withdrawal (up to 36 weeks)
period C – double-blind retreatment study based on original randomisation in period A (16 weeks)
period D – long-term follow-up (up to 52 weeks).
The double-blind RCT (period A) recruited paediatric patients (aged 4–17 years, weighing ≥ 13 kg) with severe chronic psoriasis from 42 centres across 13 countries. Severe chronic psoriasis was defined as failure to respond to topical therapy, requiring systemic treatment to control disease, and one of the following: (1) sPGA of ≥ 4, (2) BSA involvement of > 20%, (3) very thick lesions with BSA involvement of > 10%, (4) PASI score of > 20, or (5) PASI score of > 10 plus one of the following: (i) active psoriatic arthritis unresponsive to non-steroidal anti-inflammatory drugs, (ii) clinically relevant facial involvement, (iii) clinically relevant genital involvement, (iv) clinically relevant hand and/or foot involvement, or (v) CDLQI score of > 10.
In total, 114 participants were randomised: 38 to standard-dose adalimumab (subcutaneous; initial dose of 0.8 mg/kg up to a maximum of 40 mg, followed by 0.8 mg/kg every other week); 39 to low-dose adalimumab (subcutaneous; initial dose of 0.4 mg/kg up to a maximum of 20 mg, followed by 0.4 mg/kg every other week); and 37 to methotrexate (orally; initial dose of 0.1 mg/kg up to a maximum of 7.5 mg, followed by a weekly dose of up to 0.4 mg/kg, up to a maximum dose of 25 mg/week). To maintain blinding, participants allocated to adalimumab received placebo tablets and participants allocated to methotrexate received a placebo injection according to the adalimumab schedule. As methotrexate is a folic acid antagonist, all participants received folic acid (0.8–1.0 mg/day) as a dietary supplement (to maintain study blinding).
Previous therapy received by trial participants included topical therapy (100%), phototherapy (52%), non-biological systemic therapy (30%) and biological therapy (10%; all etanercept).
Risk-of-bias assessment
The risk of bias for the trial was low for most domains, with appropriate methods used for the allocation of participants, blinding, handling of missing data and reporting of outcomes (on the basis of information reported in the CSR;45
). Baseline characteristics were mostly balanced across treatment groups, with the exception of percentage of males, which appeared to be lower in the methotrexate arm. It should be noted that only six of the 114 children randomised were aged < 7 years at recruitment, all of whom were randomised to the low-dose adalimumab group. This means that, despite adalimumab having a marketing authorisation in children aged ≥ 4 years, this particular trial does not provide any comparative efficacy data on the licensed standard dose of adalimumab in children aged 4–6 years.
Risk-of-bias assessment using the Cochrane risk-of-bias tool for the adalimumab trial (M04-717) (period A)
In total, 16 of the 114 participants received the wrong medication. Regulatory documents indicate that the incidence of the error ‘wrong medication’ occurred at single time points and was unlikely to have affected the results of the study.45 A small number of patients (n < 5) across the three treatment arms received topical therapies during the randomised period, despite it being prohibited under the trial protocol, although this is unlikely to have had a substantial impact on the efficacy estimates.
Primary efficacy end points for the randomised controlled period were a ≥ PASI 75 response at week 16 and a sPGA rating of ‘cleared’ or ‘minimal’ (0 or 1) at week 16. Secondary outcomes included PASI 50, 90 and 100 responses, a PGA score of 0 and CDLQI and PedsQL scores.
Participants were evaluated at all visits for worsening of psoriasis. Up to and including the week 8 visit, participants were eligible for ‘early escape’ if they met one of the following criteria: (1) PASI scores increased by 50% at week 4 relative to baseline or (2) PASI scores increased by 25% relative to baseline and by ≥ 4 points at each of two consecutive study visits (prior to or at week 8). After week 8, participants were to continue in the trial until the week 16 visit.
Participants entering ‘early escape’ were permitted to enter a longer-term observational study period (period D; see Period D: long-term follow-up) in which they received open-label adalimumab at a dose of 0.8 mg/kg every other week (up to a maximum of 40 mg).
Primary efficacy analyses were conducted in the intention-to-treat (ITT) population (i.e. all randomised participants). Participants with missing or incomplete data at week 16 (including those entering ‘early escape’) were imputed to be non-responders for categorical variables (non-responder imputation method) and had their last observation carried forward for continuous variables. Analyses using per-protocol and ‘as observed’ data were also reported in the CSR.45 The safety analysis was conducted in the safety population (i.e. all participants who received at least one dose of the study medication).
Efficacy of adalimumab at 16 weeks
The absolute and relative results for PASI, sPGA, CDLQI and PedsQL outcomes at week 16 are shown in and .
Results of key outcomes in the adalimumab trial (M04-717) at 16 weeks
Relative risks of key outcomes in the adalimumab trial (M04-717) at 16 weeks
Psoriasis Area and Severity Index response
(Confidential information has been removed) and PASI 75 response rates at 16 weeks were significantly greater for standard-dose adalimumab (0.8 mg/kg) than for methotrexate [(confidential information has been removed) 58% vs. 32% respectively)]. Low-dose adalimumab (0.4 mg/kg) did not show a statistically significant improvement over methotrexate for these outcomes [(confidential information has been removed) and 44% vs. 32% respectively]. PASI 90 response rates did not differ significantly between the three treatment arms ().
Psoriasis Area and Severity Index and sPGA responses by age subgroups at 16 weeks
(Confidential information has been removed.)
Physician Global Assessment
The proportion of participants achieving a sPGA score of 0 or 1 (‘clear’ or ‘minimal’) at 16 weeks was greater for standard-dose adalimumab than for low-dose adalimumab or methotrexate (61% vs. 41% vs. 41% respectively), although this difference was not statistically significant.
(Confidential information has been removed.)
Quality of life
Two HRQoL measures, CDLQI and PedsQL, were reported at 16 weeks. All three treatment groups showed improvements from baseline in the dermatology-specific quality-of-life measure (CDLQI), exceeding the published minimal clinically important difference (MCID) of a 2.5-point change from baseline.48 However, these improvements were similar across the three treatment groups, with no significant difference between either dose of adalimumab or methotrexate (6.6 for adalimumab 0.8 mg/kg vs. 4.9 for adalimumab 0.4 mg/kg vs. 5.0 for methotrexate respectively).
Unlike the CDLQI, improvements on the generic HRQoL measure (PedsQL) significantly favoured both doses of adalimumab over methotrexate (mean changes of 10.8 and 9.5 for standard- and low-dose adalimumab, respectively, vs. 1.9 for methotrexate). The mean changes in the adalimumab groups both exceeded the published MCID of 4.4 for the PedsQL.26
It is unclear why PedsQL scores would increase in the absence of dermatology-related quality-of-life benefits as measured by the CDLQI. However, both mean and median PedsQL scores at baseline were noticeably higher in the methotrexate arm than in the adalimumab treatment arms (see ) and so the observed PedsQL change scores in the adalimumab arms may be overestimates because of regression to the mean.89
Longer-term efficacy of adalimumab
Period B: withdrawal
(Confidential information has been removed.)
Period C: retreatment
(Confidential information has been removed.)
Reported PASI responses during the retreatment phase (week 16 of period C)
(Confidential information has been removed.)
Period D: long-term follow-up
(Confidential information has been removed.)
Reported PASI responses during the long-term follow-up phase (week 52 of period D)
Safety of adalimumab
Adverse events at 16 weeks
Adverse event rates were comparable among the three treatment groups (). Three serious adverse events (SAEs) considered unrelated to treatment (hand fracture, gastrointestinal infection from food poisoning and agitation as a result of alcohol consumption) were reported, all of which occurred in participants receiving 0.4 mg/kg of adalimumab. One participant in the same treatment arm withdrew because of an AE (moderate psoriasis flare).
Reported safety outcomes in the adalimumab trial (M04-717) at week 16
Longer-term safety of adalimumab
shows that the overall numbers of AEs during patient follow-up across all four study periods were similar across treatment arms. A total of nine SAEs were reported in six participants. In terms of episodes per 100 patient-years, the total rate of SAEs was 5.9 for all participants ever treated with 0.8 mg/kg of adalimumab from the first dose of 0.8 mg/kg adalimumab and 7.4 for all participants treated with adalimumab (0.4 mg/kg and 0.8 mg/kg) from the first dose of 0.8 mg/kg adalimumab.
Reported safety outcomes in the adalimumab trial (M04–717) in the different follow-up periods
One SAE (haemorrhagic ovarian cyst) occurred in period B in a participant who had been initially randomised to 0.8 mg/kg of adalimumab.
Five SAEs occurred during period D, including one death from an accidental fall, one tendon injury in a participant receiving 0.4 mg/kg of adalimumab, one maculopapular rash in a participant receiving 0.8 mg/kg of adalimumab, one case of chest pain in a participant randomised to methotrexate but receiving 0.8 mg/kg of adalimumab and one case of eye naevus in a participant receiving 0.8 mg/kg of adalimumab. All SAEs were considered by investigators to be unrelated or probably unrelated to the study drug with the exception of the case of eye naevus, which was assessed as being possibly related.
In addition to the participant who discontinued treatment because of a moderate psoriasis flare in period A, one participant initially randomised to methotrexate but receiving 0.8 mg/kg of adalimumab during period D discontinued treatment because of severe urticaria.
The rate of all infections reported by participants receiving 0.8 mg/kg of adalimumab was 170.4 episodes per 100 patient-years. Only two events of tuberculosis occurred, both during period D.
Summary of the efficacy and safety of adalimumab
There was evidence from one 16-week RCT comparing adalimumab with methotrexate in children and young people with severe chronic psoriasis.
This trial did not provide comparative evidence for children aged 4–6 years.
Adalimumab at the licensed dose of 0.8 mg/kg (up to 40 mg) leads to significantly greater responses than methotrexate for the outcomes of PASI 50 and PASI 75, but not for the outcome of PASI 90.
PGA 0/1 response rates were higher for 0.8 mg/kg of adalimumab than for methotrexate, although the difference was not statistically significant.
The benefits of half-dose adalimumab were not statistically greater than those observed for methotrexate.
Evidence on quality of life was inconsistent across different measures, possibly because of baseline imbalances on the PedsQL.
(Confidential information has been removed.)
In children and young people, adalimumab does not appear to be associated with an increase in adverse effects relative to methotrexate over 16 weeks, (confidential information has been removed).
However, because of the small numbers of observed participants, the possibility of rare AEs cannot be entirely excluded.
Efficacy and safety of etanercept
One multicentre RCT (20030211) comparing etanercept with placebo met the selection criteria for the review. Data on short-term safety and efficacy (blinded period) were available from published peer-reviewed journal papers,48–53 conference proceedings54–60 and regulatory documents.61–71
This double-blind RCT recruited children aged between 4 and 17 years from 42 sites in the USA and Canada who had stable, moderate-to-severe plaque psoriasis at screening. Moderate to severe plaque psoriasis was defined as a PASI score of ≥ 12 (PASI scores range from 0 to 72, with higher scores indicating a worse condition); a sPGA of ≥ 3 (in which 0 indicates clear and 5 indicates severe psoriasis) and psoriasis involvement of ≥ 10% of the BSA; a history of psoriasis for ≥ 6 months; and previous or current treatment with phototherapy or systemic psoriasis therapy (e.g. methotrexate, ciclosporin or retinoids) or psoriasis considered by the investigator as poorly controlled with topical therapy.
Within each age stratum, participants were randomised in a 1 : 1 ratio to either 0.8 mg/kg of etanercept once weekly up to a maximum dose of 50 mg or placebo.
The primary outcome measure used in the RCT was the PASI 75 response at week 12. The secondary outcome measures were PASI 50 response, PASI 90 response, clear or almost clear status on the sPGA and percentage improvement from baseline in the CDLQI at week 12.
A total of 264 participants were screened and 211 children were randomised to etanercept (n = 106) or placebo (n = 105). At baseline, both groups were similar in terms of age and sex, BSA and PASI and PGA scores, although the placebo group had a slightly higher proportion of patients with psoriatic arthritis (13% vs. 5%). There was no previous use of biological therapy in either group (see ). It should be noted that only 19 children included in the study (9.0%) were aged < 8 years and only nine (4.3%) were aged < 6 years.
At or after week 4, participants with a > 50% increase or an absolute increase of ≥ 4 points in the PASI score from baseline were allowed to enter an ‘escape’ arm to receive open-label etanercept every week up to week 12. During this initial 12-week comparative period, a higher number of participants from the placebo group (n = 27/105) than the etanercept group (n = 5/106) entered the early escape arm. Participants who entered the escape arm were recorded as non-responders at the time that they entered the escape arm. Data for those participants from before they entered the escape arm were not changed. For participants who had missing data, their missing data were imputed as non-responses but their existing data were included as observed.
Risk-of-bias assessment
The trial had a low overall risk of bias for most domains, with appropriate methods used for randomisation, handling of missing data and reporting of outcomes (). The study was described as ‘double-blinded’, although the methods used to achieve blinding were not described.
Risk-of-bias assessment using the Cochrane risk-of-bias tool for the etanercept trial (20030211)
Efficacy of etanercept at week 12
Data on the treatment response outcomes were available from publications and regulatory documents.49–70,87,89 PASI and PGA scores are reported in and .
Reported treatment response and HRQoL outcomes in the etanercept trial (20030211) at week 12
Relative risks of key outcomes in the etanercept trial (20030211) at week 12
Psoriasis Area and Severity Index response
Psoriasis Area and Severity Index 50, 75 and 90 responses in the etanercept group were 74.5%, 56.6% and 27.4% respectively. Response rates for the placebo group were 22.9%, 11.4% and 6.7%. When translated into relative risk (RR) values, the etanercept group had a significantly higher probability of achieving PASI 50, 75 and 90 responses, with RRs of 3.26 [95% confidence interval (CI) 2.26 to 4.71], 4.95 (95% CI 2.84 to 8.65) and 4.10 (95% CI 1.88 to 8.95) respectively.
Physician Global Assessment
The proportion of participants achieving a PGA score of 0 or 1 (‘clear’ or ‘minimal’) at 12 weeks was significantly greater in the etanercept group than in the placebo group (52.8% vs. 13.3%), equating to a RR of 3.96 (95% CI 2.36 to 6.66).
Quality of life
Data for two HRQoL measures, CDLQI and PedsQL, were available at 12 weeks (see ). Both the etanercept group and the placebo group showed improvements from baseline in CDLQI scores, exceeding the published MCID of a 2.5-point change from baseline,48 although the improvement in the etanercept group was statistically significantly greater than that in the placebo group (mean difference 2.3, 95% CI 0.85 to 3.74).
Both treatment groups also showed improvements on the PedsQL, although for the placebo group this fell below the published MCID of 4.4. The mean change in PedsQL score from baseline, although favouring etanercept, was not statistically significantly different between the treatment groups (mean difference 3.0, 95% CI –0.87 to 6.87).
Subgroup outcomes
Age-based subgroup analysis results of PASI responses for the etanercept trial (20030211) were available (). A higher proportion of the etanercept treatment group than the placebo group achieved PASI 50, 75 and 90 responses in all age categories. Imputation of treatment failure for participants entering the early escape arm reduced the magnitude of the difference between treatments, although these differences remained formally statistically significant for all comparisons, with the exception of PASI 90, which was of borderline statistical significance (p = 0.054).
Subgroup PASI responses at week 12 (published results)
Longer-term efficacy of etanercept
Weeks 12–36: open-label etanercept treatment
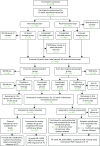
At the end of the 12-week double-blind period a total of 208 participants (105 and 103 of the original etanercept and placebo groups respectively) entered an open-label treatment phase (i.e. all were treated with etanercept) and were followed up until week 36.
Patients who did not achieve a PASI 50 response at week 24 were given the option to discontinue the study or enter the incomplete responder arm. Participants in the incomplete responder arm had the option to receive topical psoriasis therapy according to the standard of care in addition to receiving open-label etanercept ().
Short-term and long-term participant follow-up flow chart for the etanercept trial.
By weeks 24 and 36 (i.e. after 12 and 24 weeks of open-label etanercept respectively), participants who were originally randomised to placebo during the double-blind period achieved similar PASI and PGA responses as participants receiving etanercept throughout ().
Results of key outcomes in the etanercept trial (20030211) at 12 and 36 weeks
Weeks 36–48: re-randomised ‘withdrawal–retreatment’ period
At week 36, 138 patients who had achieved a PASI 50 response at week 24 or a PASI 75 response at week 36 were randomised in a 1 : 1 ratio to receive either etanercept or placebo in a double-blinded fashion. Patients were followed up for a further 12 weeks until week 48.
During the follow-up, 42 participants from the ITT population (29/69 and 13/69 from the placebo and etanercept arms respectively) lost their PASI 75 response and so were allocated to receive etanercept in an open-label fashion until week 48.
Overall, 52 out of 65 participants (80%) who received etanercept throughout the withdrawal–retreatment period maintained a PASI 75 response and 85% of those re-randomised to placebo and who did not lose a PASI 75 response during follow-up retained their response at week 48. Only 36% of those who were retreated with open-label etanercept after losing a PASI 75 response on placebo had regained a response by week 48 (). The use of PASI 75 response as both a retreatment rule and as an outcome makes these results difficult to interpret; however, a relatively high rate of late crossover from placebo to etanercept could partly explain a lack of response on PASI and PGA measures among these participants.
Results of key outcomes in the etanercept trial (20030211) at week 48 (observed data)
Weeks 48–312 (20050111)
In total, 194 participants completed 48 weeks of follow-up in the etanercept trial (20030211) (57 participants who received etanercept and topical therapy starting from the open-label treatment phase, 95 participants who were randomised to the etanercept and placebo arms and who continued to receive the blinded drug and 42 participants who were randomised to either etanercept or placebo but who did not achieve a PASI 75 response and who were retreated with etanercept until the end of the study).
Of the 194 participants who completed the etanercept trial (20030211), 182 were enrolled in an open-label extension study (20050111)66 to establish the long-term safety of etanercept. Participants received 0.8 mg/kg of etanercept (up to a maximum dose of 50 mg) subcutaneously once weekly for a further 264 weeks. In total, 63 participants (34.6%) completed 264 weeks of follow-up.
During the 264 weeks of further follow-up, the probability of achieving a PASI 50, 75 and 90 response was similar across all of the outcome recording points (). However, it should be noted that, by week 264, 63.6% of the participants (115/181) had withdrawn from the study and the reasons for withdrawal were unavailable.
Reported efficacy outcomes during the long-term follow-up period (20050111)
Safety of etanercept
Adverse events at 12 weeks
The number of AEs reported during the 12-week randomised phase was similar for etanercept and placebo (68 vs. 62). There were 50 infections and seven injection site reactions in the etanercept group, compared with 33 infections and five injection site reactions in the placebo group. Although the difference in rate of infections fell short of formal statistical significance, there were noticeably more infections in the etanercept group (47.2% vs. 31.4%; p = 0.0683). One participant in the etanercept group withdrew because of an adverse effect (no further details available). No SAEs were observed during the 12-week randomised phase ().
Reported safety outcomes in the etanercept trial (20030211) at week 12
Adverse events at weeks 12–36
Up to week 36, 282 infections were reported during treatment with etanercept (238.18 events per 100 person-years). The most common infections were upper respiratory tract infection, nasopharyngitis, influenza, pharyngitis, streptococcal pharyngitis and viral upper respiratory tract infection (59.97, 33.79, 16.05, 15.20, 8.45 and 8.45 events per 100 participant-years respectively). During the same period, a single serious non-infectious AE (benign ovarian mass) and one serious case of gastroenteritis with dehydration were observed ().
Reported safety outcomes in the etanercept trial (20030211) up to week 36
Over the same time period, 2.4% of participants (5/208) withdrew because of AEs: three participants withdrew because of non-infectious AEs (psoriasis, atopic dermatitis and muscle cramps) and two participants withdrew because of infections (pneumonia and skin infection).
Adverse events at weeks 48–312
A total of 161 participants (89.0%) reported at least one AE up to week 264 of the follow-up study (20050111). Seven participants (3.9%) reported a SAE, with each participant reporting a single event: anxiety, cellulitis, infectious mononucleosis, postoperative intestinal obstruction, osteonecrosis and a thyroid cyst, with the seventh participant undergoing an elective abortion (). Of the seven SAEs, only the cellulitis infection was considered by the investigator to be related to etanercept treatment.
Reported safety outcomes in the etanercept trial (20050111) up to week 264
Six participants (3.3%) withdrew from the study because of either an infectious or a non-infectious AE. Two participants withdrew because of Crohn’s disease and one participant each withdrew because of glomerulonephritis (secondary to infection), psoriasis, sinusitis and nerve paralysis. The case of glomerulonephritis and one of the cases of Crohn’s disease were considered to be related to treatment.
No SAE led to study withdrawal. No opportunistic infections or deaths occurred during the study and no malignancies were reported.
Summary of the efficacy and safety of etanercept
One multicentre RCT (20030211) compared etanercept with placebo in children aged 4–17 years with moderate to severe plaque psoriasis.
Relatively few young children (9% aged < 8 years; 4.3% aged < 6 years) were included in the study.
At 12 weeks etanercept was significantly more effective than placebo in improving the severity of plaque psoriasis based on PASI 50, 75 and 90 and PGA cleared or minimal scores.
Improvements in HRQoL were larger for etanercept than for placebo, but reached statistical significance only for the CDLQI.
Adverse events rates were similar between the etanercept group and the placebo group at 12 weeks, with no SAEs observed for either treatment. However, the observed higher rate of infections among participants receiving etanercept was of borderline statistical significance.
A subsequent open-label extension study followed up participants for up to 6 years from entry into the original RCT. The proportions of PASI and PGA responders were stable over time, although only 36% of participants were available at the last follow-up point. The proportion of participants withdrawing because of lack of efficacy is unknown. These longer-term uncontrolled observational response data may therefore overestimate the efficacy of etanercept.
Withdrawals because of AEs were infrequent and no deaths or malignancies were observed up to 264 weeks of additional follow-up.
Efficacy and safety of ustekinumab
One multicentre RCT (1275PSO3006; CADMUS) comparing standard and half-standard dosages of ustekinumab with placebo met the selection criteria for the review. Data on safety and efficacy (blinded period) were available from one peer-reviewed journal paper,72 a conference abstract,73 regulatory documentation74 and a CSR provided by the manufacturer.88
The CADMUS RCT was a double-blind, placebo-controlled study in adolescent participants (aged 12–17 years) who had had a diagnosis of moderate to severe plaque psoriasis for ≥ 6 months, which was conducted at multiple sites in Europe (Belgium, France, Germany, Hungary, Portugal, Russian Federation, Sweden, Ukraine and the UK) and Canada. Moderate-to-severe disease was defined as a PASI score of ≥ 12, a PGA score of ≥ 3 and BSA involvement of ≥ 10%.
A total of 157 participants were screened, of whom 110 were eligible and randomised (37 participants to placebo, 37 participants to the half-standard dosage of ustekinumab and 36 participants to the standard dosage of ustekinumab). The standard dosage of ustekinumab was 0.75 mg/kg for participants weighing ≤ 60 kg, 45 mg for participants weighing 60–100 kg and 90 mg for participants weighing > 100 kg. The half-standard dosage of ustekinumab was 0.375 mg/kg for participants weighing 60–100 kg and 45 mg for participants weighing > 100 kg. Randomisation was stratified by investigational site and baseline weight (≤ 60 kg or > 60 kg).
The study had three periods:
Controlled period (0–12 weeks): participants received either ustekinumab (full dose or half dose) or placebo. In the ustekinumab groups, participants were allowed to escape early at week 8 and receive moderate- to high-potency topical steroid preparations up to week 12 if their PASI scores increased by ≥ 50% from baseline. However no participants entered the escape route during this period.
Placebo crossover and active treatment period (12–52 weeks): participants randomised to placebo during the controlled period were allowed to cross over to the full or half-standard dose of ustekinumab at week 12.
Follow-up period (52–60 weeks): participants continued to be followed for safety analysis.
To preserve blinding, participants in the half-standard-dosage and standard-dosage groups received ustekinumab at week 0 and week 4 followed by doses every 12 weeks until week 40. Participants in the placebo group also received placebo at week 0 and week 4 and crossed over to receive either the half-standard dosage or standard dosage of ustekinumab at week 12 or week 16, followed by 12 weekly doses of either a half-standard dosage or standard dosage of ustekinumab, with the last dose at week 40. All participants were followed up for efficacy up to week 52 and for safety up to week 60.
The primary outcome measure was the proportion of participants who achieved a sPGA score of ‘cleared’ or ‘minimal’ at week 12. Data from all randomised participants were analysed according to their assigned treatment group. Participants who met treatment failure criteria prior to week 12 or who entered the early escape arm were considered non-responders at week 12. In addition, participants who had a missing PGA score at week 12 were considered as not achieving the primary end point at week 12.
The secondary outcome measures were PASI 50, 75 and 90 response at week 12 based on all randomised participants and changes from baseline in CDLQI score at week 12 based on efficacy-evaluable participants.
Risk-of-bias assessment
Based on the Cochrane risk-of-bias assessment tool,34 the CADMUS trial double-blind period had a low risk of bias: appropriate randomisation and blinding techniques were implemented, no obvious difference in baseline characteristics between treatment arms was apparent, missing data were handled appropriately and all protocol-stated outcome measures were reported ().
Risk-of-bias assessment using the Cochrane risk-of-bias tool for the ustekinumab trial (CADMUS)
Efficacy of ustekinumab
Efficacy at week 12
Data on treatment response (PASI and sPGA) and HRQoL (CDLQI and PedsQL) outcomes for the CADMUS RCT are presented in and .
Results of key outcomes in the ustekinumab trial (CADMUS) at week 12
Relative risks of key outcomes in the ustekinumab trial (CADMUS) at week 12
Physician Global Assessment
Significantly greater proportions of participants in the standard-dosage and the half-standard-dosage groups (69.4% and 67.6% respectively) than in the placebo group (5.4%) achieved a PGA score of cleared (0) or minimal (1) at week 12. The proportions of participants who achieved a PGA score of cleared (0) were also higher in the standard-dosage and half-standard-dosage groups (47.2% and 32.4% respectively) than in the placebo group (2.7%). The RRs for these outcomes are shown in .
Psoriasis Area and Severity Index response
Higher proportions of participants in the standard-dosage and the half-standard-dosage groups than in the placebo group achieved PASI 50, 75 and 90 responses. For example, 80.6% and 78.4% of the standard-dosage and half-standard-dosage groups, respectively, achieved a PASI 75 response at week 12 whereas only 10.8% of the placebo group achieved the same PASI 75 response (see ). The RR values also show that both ustekinumab dosage groups had significantly higher probabilities of achieving the PASI 50, 75 and 90 responses than the placebo group (see ).
Health-related quality of life
Changes from baseline in CDLQI score were significantly greater in both the standard-dosage and half-standard-dosage groups (mean of –6.7 and –5.6 respectively) than in the placebo group (–1.5). Although both ustekinumab treatment groups showed improvements from baseline in the CDLQI that exceed the published MCID of a 2.5-point change from baseline, this was not the case for the placebo group. The mean difference values indicate that CDLQI changes were significantly greater for both ustekinumab dosage groups than for the placebo group (mean difference: standard dosage 5.2, 95% CI 2.96 to 7.44; half-standard dosage 4.1, 95% CI 1.7 to 6.5; see ).
Participants in both the standard-dosage group and the half-standard-dosage group showed significantly larger improvements in the PedsQL total scale scores from baseline (mean 8.03 and 10.81 respectively) than participants in the placebo group (mean 3.35). The mean changes for the half-standard dosage and standard dosage were above the published MCID of 4.4 whereas the mean change in the placebo group was below the MCID. Mean differences at 12 weeks indicate that the standard-dosage and half-standard-dosage groups had a significantly higher improvement in PedsQL score than the placebo group (mean difference: standard dosage 8.9, 95% CI 2.46 to 15.34; half-standard dosage 7.6, 95% CI 2.16 to 13.04).
Subgroup efficacy outcomes
Subgroup efficacy results for the PASI 75 and PGA 0 or 1 outcomes were available from the CSR88 (). (Confidential information has been removed.)
Subgroup efficacy outcomes in the ustekinumab trial (CADMUS) at week 12
Longer-term efficacy of ustekinumab
Psoriasis Area and Severity Index response
Among participants continuing ustekinumab treatment, PASI responses observed in week 12 appeared to be sustained at week 52, with few participants lost to follow-up (one participant was lost from the standard-dose arm and two from the half-standard-dose arm) ().
Results of key outcomes in the ustekinumab trial (CADMUS) at week 52
Participants who were randomised to placebo and who crossed over to the standard ustekinumab dosage (0.75 mg/kg) achieved better PASI responses than those who were randomised to placebo and crossed over to the half-standard dosage of ustekinumab (0.375 mg/kg) (see ).
Physician Global Assessment
A similar pattern of responses was seen for the sPGA, with similar response rates at week 52 as at week 12 among participants continuing ustekinumab treatment and a large improvement between weeks 12 and 52 for participants crossing over to active treatment from placebo (see ).
Safety of ustekinumab
Adverse events at week 12
The percentage of participants reporting AEs did not differ significantly between the ustekinumab groups (44.4% standard-dosage group and 51.4% half-standard-dosage group) and the placebo group (56.8%) (). No SAEs were reported in the standard-dosage group or the placebo group, whereas one participant in the half-standard-dosage group was hospitalised for worsening of psoriasis (see ).
Reported safety outcomes in the ustekinumab trial (CADMUS) at week 12
One participant in the standard dosage group had a mild injection site reaction. There were no incidences of serious infection, tuberculosis, malignancy or withdrawals because of AEs during the initial 12-week treatment period (see ).
Discontinuation up to week 40
Up to week 40, 8.2% of the participants (9/110) discontinued the trial. The most common reasons for discontinuation were a lack of efficacy and AEs (). Five participants (13.5%) who were originally randomised to the half-standard-dosage group discontinued the trial compared with two participants (5.6%) in the standard-dosage group. Two patients who crossed over from placebo to the half-standard dosage of ustekinumab withdrew because of AEs.
Reported number of participants discontinuing ustekinumab treatment (CADMUS) up to week 40
Adverse events up to week 60
Up to week 60, 81.8% of participants (90/110) in the ustekinumab combined group reported one or more AEs (). Of the 74 participant-recorded infections, 18 (24%) were considered reasonably related to ustekinumab treatment.
Reported safety outcomes in the ustekinumab trial (CADMUS) up to week 60
In total, 5.5% of participants (6/110) in the ustekinumab combined group (five participants in the half-standard-dosage group and one participant in the standard-dosage group) reported SAEs (see ). Four (one because of worsening of psoriasis) of the 110 participants in the ustekinumab combined group discontinued the trial because of an AE by week 60.
Summary of the efficacy and safety of ustekinumab
Both the standard dosage and the half-standard dosage of ustekinumab were significantly more effective than placebo in improving the severity of plaque psoriasis based on PASI 50, 75 and 90 responses, sPGA cleared or minimal response and sPGA cleared response at 12 weeks.
Both ustekinumab dosages also led to significantly greater improvements in HRQoL (CDLQI and PedsQL) at 12 weeks than placebo.
Among participants originally allocated to ustekinumab, PASI and PGA effects observed at 12 weeks appeared to be largely sustained at 52 weeks, with few withdrawals because of lack of efficacy.
Participants originally allocated to placebo showed substantial improvements in PASI and PGA responses at 52 weeks after crossing over to ustekinumab treatment at week 12. There was some indication that the gains were greater among those who received the standard dosage of ustekinumab than among those who received the half-standard dosage.
There were no notable differences between the ustekinumab group and the placebo group in terms of short-term and longer-term AEs, although the number of observations was small and the longest follow-up time was only 60 weeks. Few participants withdrew because of adverse effects.
Additional observational evidence
Retrospective case series
Two retrospective case series reported the use of adalimumab, etanercept and ustekinumab from 4 to 165 weeks in children with moderate to severe psoriasis.76,77 The key characteristics and results from these studies are reported in .
Retrospective case series of adalimumab, etanercept or ustekinumab in children and young people with psoriasis
Garber et al.76 reported a retrospective chart review of 27 participants (19 males, 8 females) attending a single US general dermatology clinic from 2008 to 2014. Insufficient details were reported to establish how many patients received more than one biologic over this period. Clearance rates [defined as 99% reduction on the Simple Measure for Assessing Psoriasis Severity or (S-MAPA)] were reported (see ). No SAEs were reported. Although the authors concluded that the use of adalimumab, etanercept and ustekinumab is safe in paediatric psoriasis and that these treatments are efficacious, this study provides insufficient data on the efficacy or safety profiles of these agents in practice.
Klufas et al.77 similarly reported a retrospective case series evaluating 51 children with moderate to severe psoriasis treated with systemic therapies for AE occurrence and PGA-measured disease response. For all biologics (alone or in combination with methotrexate), mean PGA values fell at 5–7 months’ follow-up. In total, 29 AEs were reported in relation to 80 treatment data points (some patients received more than one biologic); most were minor subjective side effects, with no infections or SAEs reported. Again, limitations in the sample size and study design preclude strong inferences being drawn from these data.
Registry data
Published findings or papers detailing study design were identified for 16 registries. Information on biological drug safety in their psoriasis cohorts was published by nine registries, with 11 articles on biological efficacy and nine articles including drug survival data. This does not necessarily mean that the other registries did not record these outcomes, but they were not covered in the identified literature output.
Registry data for children
Further screening was carried out to find registry publications making explicit reference to children with psoriasis and specifically providing information on the survival of biological treatment. Two registries (Child-CAPTURE and DERMBIO) were found to include children with psoriasis who were treated with biologics. Child-CAPTURE (the Netherlands) contained seven children treated with etanercept,90 but did not differentiate between biological and non-biological therapies in drug survival analyses. We identified from the 2014 annual report91 by the DERMBIO (Denmark) registry that there are 37 children enrolled who are undergoing treatment with adalimumab, etanercept or ustekinumab, although data for this group were not reported separately. Cox regression modelling of covariates in two studies found no significant predictive relationship between patient age and drug survival, suggesting that treatment withdrawal rates among children were similar to those in adults.92,93
Wider registry data
In one 2015 DERMBIO study following 1277 (predominantly adult) psoriasis patients for up to 10 years,92 median drug survival for etanercept was 30 months (95% CI 25.1 to 34.9 months), which was significantly lower than for adalimumab (59 months, 95% CI 45.6 to 72.4 months) and ustekinumab (median not reached). Year-on-year drug survival for etanercept (estimated from the Kaplan–Meier curve) was 0.70 at 1 year, 0.53 at year 2 and 0.30 at year 5, whereas year-on-year drug survival for ustekinumab was 0.85 after 1 year, 0.78 at year 2 and 0.65 at year 5 (). Loss of efficacy was the most likely reason for drug discontinuation, but this was of greater significance proportionally for etanercept than for the other biologics analysed.
Survival of first biologic in the DERMBIO registry
Findings from the British Association of Dermatologists Biologic Interventions Register (BADBIR) were broadly similar to those seen in the Danish cohort. A study by Warren et al.94 on drug survival over 3 years in 3523 biologic-naive patients found that 77% of patients remained on biologic treatment over the first year, falling to 53% by the third year. Again, there were significant differences in the treatment withdrawal rates between biologics. Ustekinumab exhibited the highest first-course survival rate at 0.89 at year 1 and 0.75 at year 3. Adalimumab showed the highest survival of the anti-TNF-α drugs, at 0.79 at year 1 and 0.59 at year 3. Disregarding the very small population of patients on infliximab, etanercept was consistently the worst-performing drug in terms of treatment withdrawal, with a 1-year survival rate of 0.70, dropping to 0.40 at 3 years (). Etanercept was also found to be a significant predictor of discontinuation of therapy because of loss of efficacy. Other significant predictors of treatment withdrawal were female sex, smoking status and a higher baseline Dermatology Life Quality Index (DLQI) score.
Survival of first biologic in the BADBIR
Overview of the randomised controlled trial results
Despite differences in inclusion criteria, the relative lack of younger children in the adalimumab and etanercept trials meant that the median age of children across the three trials did not differ greatly. Similarly, measures of disease duration and the component measures of severity did not appear to differ markedly between the three trials. Few participants in any trial had previous experience of biological treatment.
The biologics and their respective comparators in the relevant RCTs in children and young people are summarised in .
Summary of the biologics and their comparators based on RCTs
There were no head-to-head comparative data available for the three biologics. In addition, although the etanercept and ustekinumab trials had a placebo as a common comparator, the adalimumab trial used methotrexate as a comparator.
shows the relative effects for all three biologics from the three RCTs. However, an implicit comparison is not useful for the purposes of the decision-analytic modelling required for the economic evaluation. Chapter 4 therefore describes a formal evidence synthesis to inform the relative efficacy of these interventions.
Relative risks of PASI outcomes for biological therapy trials in children and young people